3192
Views & Citations2192
Likes & Shares
Sturge-Weber Syndrome (SWS) is a rare disorder characterized by the association of a facial birthmark called a port-wine birthmark, neurological abnormalities and eye abnormalities such as glaucoma. SWS can be thought of as a spectrum of disease in which individuals may have abnormalities affecting all three of these systems (i.e., brain, skin and eyes) or only two or only one. Consequently, the specific symptoms and severity of the disorder can vary dramatically from one person to another. Symptoms are usually present at birth (congenital), yet the disorder is not inherited and does not run in families. Some symptoms may not develop until adulthood. SWS are caused by a somatic mutation in the GNAQ gene. This mutation occurs randomly (sporadically) for no known reason.
Keywords: Sturge-weber syndrome, Genetic mutations, GNAQ gene, Glaucoma, Seizure
OVERVIEW OF STURGE-WEBER SYNDROME
Sturge-Weber syndrome is a rare genetic disorder that affects the development of certain blood vessels and causes disorders in the brain, skin and eyes. Sturge-Weber syndrome has three main features: congenital red or pink congestion on the face, a brain disorder known as leptomeningeal angioma and increased intraocular pressure (glaucoma). It should be noted that not all people with Sturge-Weber syndrome have these three characteristics together [1].
SIGNS AND SYMPTOMS OF STURGE-WEBER SYNDROME
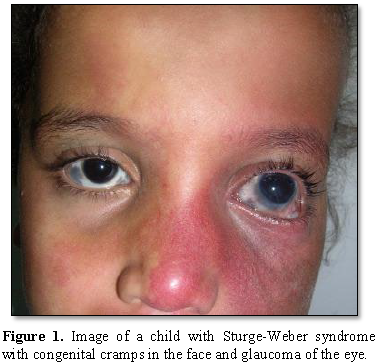
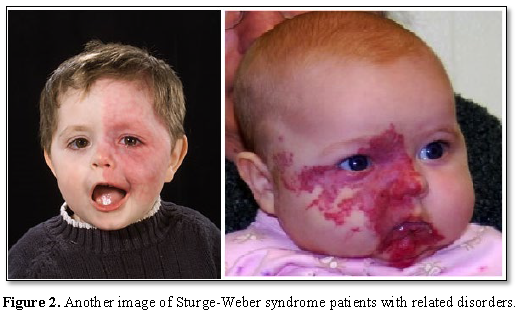
Most people with Sturge-Weber syndrome are born with pink or red congenital lunar symptoms. This type of lunar eclipse is caused by the enlargement of small blood vessels (capillaries) near the surface of the skin. The eclipse of the moon is usually initially flat and may vary from orange to dark purple. In people with Sturge-Weber syndrome, the appearance of a lunar eclipse is often seen on the forehead and eyelid skin. Eclipse usually occurs on one side of the face but can also occur on both sides of the face [1,2] (Figures 1 and 2).
In Sturge-Weber syndrome, there is usually abnormal formation and growth of blood vessels in the two thin layers of tissue that cover the brain and spinal cord. This disorder, called leptomeningeal angioma, can disrupt brain blood flow and lead to brain tissue destruction (atrophy) and calcium deposition (calcification) in the brain, sub-angiomas or cerebral arteries. Decreased blood flow due to leptomeningeal angioma can cause stroke in people with Sturge-Weber syndrome [1,3].
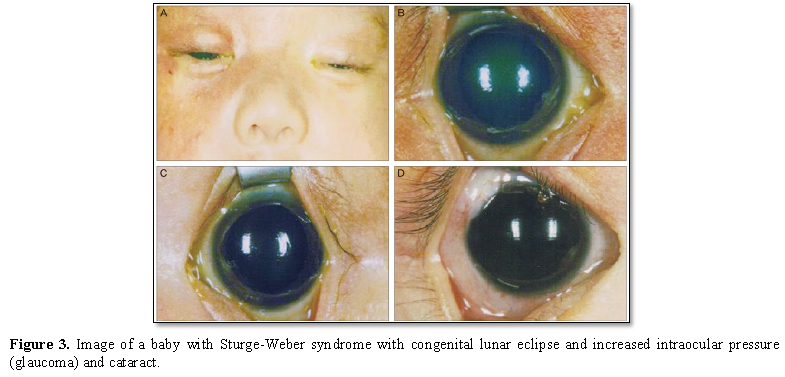
Other symptoms in Sturge-Weber syndrome include temporary muscle weakness on one side of the body (hemiparesis), visual impairments, seizures, and migraine headaches. In patients with Sturge-Weber syndrome, these symptoms usually begin at age 2. People with Sturge-Weber syndrome have different levels of cognitive functioning, from normal intelligence to intellectual disability [1,4] (Figure 3).
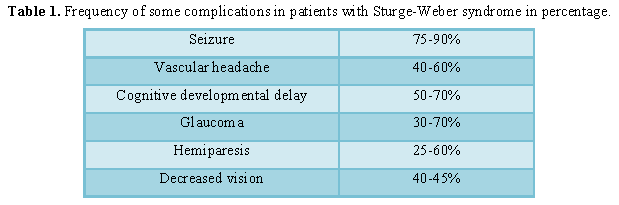
In people with Sturge-Weber syndrome, glaucoma usually occurs early in life or in childhood, which can lead to visual impairment. In some affected infants, the pressure in the eye can be very high, which can cause the eyes to become larger and shiny. People with Sturge-Weber syndrome may experience abnormal blood vessels (hemangiomas) in different parts of their eyes. When these abnormal vessels form in the back of the blood vessel network (choroid), it is known as diffuse choroidal hemangioma, which occurs in about one-third of people with Sturge-Weber syndrome. Disseminated choroidal hemangioma can cause vision loss. It is noteworthy that eye anomalies usually occur on the same side of the face as the affected skin [1,5] (Table 1).
ETIOLOGY OF STURGE-WEBER SYNDROME
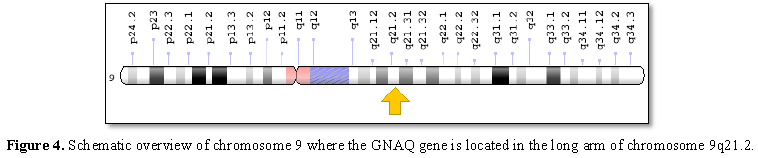
Sturge-Weber syndrome is caused by a GNAQ gene mutation located on the long arm of chromosome 9 as 9q21.2. This gene provides the necessary instructions for the synthesis of a protein called guanine nucleotide bonding (Gαq). Gαq protein is part of a protein that controls signaling pathways to control the growth and function of blood vessels [1,6] (Figure 4).
A mutation in the GNAQ gene that causes Sturge-Weber syndrome disrupts protein production and related function. Therefore, the altered Gαq protein cannot play a role in regulating signaling pathways, thereby causing anomalous signal enhancement. An increase in signal regulation disrupts the growth of blood vessels and causes excessive vascular formation in people with prenatal Sturge-Weber syndrome. Sturge-Weber syndrome does not follow any inherited pattern and occurs sporadically due to new mutations [1,6].
FREQUENCY OF STURGE-WEBER SYNDROME
Sturge-Weber syndrome is a genetic disorder estimated to have a prevalence of about 1 in 20,000 to 1 in 50,000 live births. Approximately 3 in 1,000 babies are born with a port-wine birthmark, but only approximately 6% of individuals with a port-wine birthmark on the face develop the neurological abnormalities associated with SWS. The risk increases to 26% when the port-wine birthmark is on the forehead, temple region or upper part of the face. SWS can affect individuals of any race or ethnicity [1,7].
DIAGNOSIS OF STURGE-WEBER SYNDROME
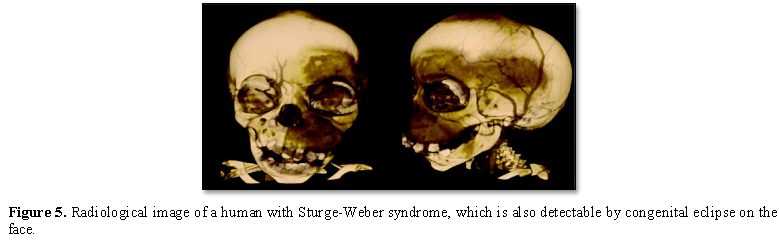
Sturge-Weber syndrome is diagnosed based on clinical findings of patients and some pathological tests. Radiological imaging techniques such as CT scans and MRIs are often used to diagnose intracranial malformations. However, the most definitive method of diagnosing Sturge-Weber syndrome is molecular genetic testing for the GNAQ gene to determine the presence of possible mutations [1,7] (Figure 5).
THE THERAPEUTIC PATHWAYS OF STURGE-WEBER SYNDROME
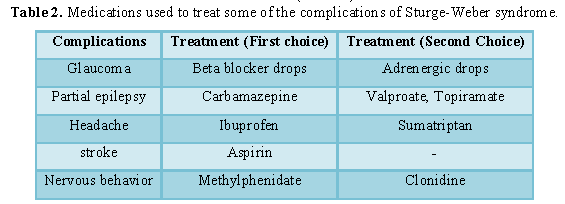
The strategy of treatment and management of Sturge-Weber syndrome is symptomatic and supportive. Laser treatment may be used to remove lunar eclipses on the face of the skin. Anticonvulsants such as phenobarbital, carbamazepine, and sodium valproate can also be useful in controlling seizures. Surgery can also be effective in treating glaucoma. Physiotherapy is effective for children with muscular weakness in Sturge-Weber syndrome. Medications such as latanoprost (xalatan) and prostaglandins can also significantly reduce IOP in patients with Sturge-Weber syndrome with glaucoma. Genetic counseling also holds a special place for all parents who want a healthy child [1,7] (Table 2).
HISTORY OF STURGE-WEBER SYNDROME
Symptoms of this syndrome were first reported by Dr. Sturge in 1879 as neurological disorders and further symptoms were described in 1922 by Dr. Weber [1,7].
DISCUSSION AND CONCLUSION
SWS may be classified as a neurocutaneous syndrome or one of the phakomatoses. Neurocutaneous syndromes or phakomatoses are broad terms for groups of disorders in which growths develop in the skin, brain, spinal cord, bones and sometimes other organs of the body. In the case of SWs these growths consist of abnormal blood vessels. Additional symptoms may occur including an abnormally large head (macrocephaly), overgrowth (hypertrophy) of the certain soft tissues underlying the port-wine birthmark, and lymphatic malformations, which are non-malignant masses consisting of fluid-filled channels or spaces thought to be caused by abnormal development of the lymphatic system. These symptoms are consistent with a related rare disorder known as Klippel-Trenaunay syndrome (KTS) and most children with these findings are classified as having KTS. Researchers are not sure whether SWS and KTS are related disorders that overlap or whether they are similar, yet distinct, rare disorders. Genes provide instructions for creating proteins that play a critical role in many functions of the body. When a mutation of a gene occurs, the protein product may be faulty, inefficient or absent. Depending upon the functions of the particular protein, this can affect many organ systems of the body. The GNAQ gene creates a protein known as Gaq that plays an important role in cell function, including the regulation of blood vessels. The specific underlying manner in which Gaq function is disrupted in individuals with SWS is not fully understood. More research is necessary to determine the exact underlying mechanisms that cause the varied symptoms of SWS. Recent data suggests that somatic mutation in GNAQ is enriched in endothelial cells which line the inside of blood vessel walls. Diagnosis can be more difficult in infants who have a port-wine birthmark, but no neurological symptoms. A complete ophthalmological exam can reveal glaucoma and other eye abnormalities potentially associated with SWS. Because of the high risk of glaucoma, complete eye examination should be performed regularly, especially in infants and young children. Follow-up examination should continue into adulthood even if results are normal through childhood. The identification of the GNAQ gene mutation will allow researchers to focus on a specific direction to better understand how SWS develops and to explore novel methods in how to treat the disorder. For example, new therapies such as drugs that specifically target the proteins and pathways associated with the GNAQ gene will be explored (targeted therapies) [1-7].
Low-dose aspirin has been used to treat individuals with SWS. Low-dose aspirin has led to a reduction in the frequency of seizures and stroke-like episodes. In some children, the decrease in seizure activity is significant. Complications have included increased bruising and gum or nose bleeding. Most reports in the medical literature suggest low-dose aspirin can safely be used in individuals with SWS and provides benefit. However, studies are needed to determine the long-term safety and effectiveness of low dose aspirin and whether this treatment improves long-term cognitive function and overall quality of life. Increasingly, low dose aspirin is being used by many centers in the treatment of SWS [1-7].
1. Asadi SH (2018) Book of Pathology in Medical Genetics. Vol 4 (M-W), Amidi Publications, Iran.
2. Comi AM, Sahin M, Hammill A, Kaplan EH, Juhász C, et al. (2015) Leveraging a Sturge-Weber Gene Discovery: An Agenda for Future Research. Pediatr Neurol 58: 12-24.
3. Comi AM (2015) Sturge-Weber syndrome. Handb Clin Neurol 132: 157-168.
4. Koenraads Y, van Egmond-Ebbeling MB, de Boer JH, Imhof SM, Braun KP, et al. (2016) Visual outcome in Sturge-Weber syndrome: A systematic review and Dutch multicentre cohort. Acta Ophthalmol 94: 638-645.
5. Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, et al. (2013) Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med 368: 1971-1979.
6. Sturge WA (1879) A case of partial epilepsy, apparently due to a lesion of one of the vasomotor centres of the brain. Trans Clin Soc Lond 12: 162.
7. Weber FP (1922) Right-sided hemi-hypertrophy resulting from right-sided congenital spastic hemiplegia, with a morbid condition of the left side of the brain, revealed by radiograms. J Neurol Psychopathol 3: 134-139.
-
 Table 1
Table 1 -
Table 2
QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Stem Cell Research and Therapeutics (ISSN:2474-4646)
- International Journal of AIDS (ISSN: 2644-3023)
- Journal of Immunology Research and Therapy (ISSN:2472-727X)
- Ophthalmology Clinics and Research (ISSN:2638-115X)
- Journal of Clinical Trials and Research (ISSN:2637-7373)
- Journal of Spine Diseases
- International Journal of Clinical Case Studies and Reports (ISSN:2641-5771)


