2495
Views & Citations1495
Likes & Shares
Cutaneous small vessel vasculitis also called Immune-complex small vessel vasculitis, can be an isolated phenomenon or presenting feature of a connective tissue disorder like SLE, or can present as a severe, life-threatening syndrome, including ANCA-associated vasculitis.
Abbreviations: SLE: Systemic Lupus Erythematosus; ANA: Anti-Nuclear Antibody; dsDNA: Double Stranded DNA; ANCA: Anti-Neutrophil Cytoplasmic Antibody; CSVV: Cutaneous Small Vessel Vasculitis
INTRODUCTION
Cutaneous small vessel vasculitis also called Immune-complex small vessel vasculitis, can be an isolated phenomenon or presenting feature of a connective tissue disorder like SLE, or can present as a severe, life-threatening syndrome, including ANCA-associated vasculitis [1].
It is important to note that the same cutaneous presentation can be seen in many other systemic small vessel vasculitides such as antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (e.g. granulomatosis with polyangitis, eosinophilic granulomatosis with polyangitis, microscopic polyangitis), cryoglobulinemic vasculitis, immunoglobulin A vasculitis (IgAV; Henoch-Schönlein purpura [HSP]) as well as other conditions such as infective endocarditis. When a patient presents with palpable purpura that reveals LCV on biopsy, the patient must be evaluated for a variety of causes and associated conditions [2].
Cutaneous vasculitis (CV) encompasses a wide and heterogeneous group of disorders characterized by the presence of necrotizing inflammatory lesions in the cutaneous blood vessels [3]. They may range from isolated CV to a severe, life-threatening syndrome, including ANCA-associated vasculitis.
In our study, the main etiological factors for CSVV were: benign isolated (40) patients, (53%), infectious etiology (14) patients, (19%), Vasculitis in background of ANA/dsDNA/ANCA positivity (12) patients, (16%), drug induced (9) patients, (12%).
The main clinical manifestations of CSVV in our study were found to be the following viz, palpable purpura in all 75 patients (100%), fever and malaise in 30 patients, (40%), ulcers in 30 patients (40%) arthritis/arthralgia in 15 patients, (20%).
60 Patients (80%) responded to systemic corticosteroids, 11patients (15%) to colchicine/dapsone and 4 patients (5%) were treated with azathioprine/methotrexate.
They were regularly followed up once in two weeks till complete recovery, although relapses occurred in 8 patients (11%).
In our study out of the sample size of 75 patients, 40 patients (53%) were found to be of the benign isolated type, which correlates very well with the previous studies wherein, benign idiopathic type accounts for 50%.
Vasculitis resulting out of infections in our study were found to be due to viral and bacterial origin Viz., Coxackie virus, M. leprae constituting 19% which correlates with the previously documented studies wherein, infections can lead on to vasculitis to an extent of 20%.
Vasculitis occurring as part and parcel of connective tissue disorders in our study amounts to 16% which is on par with the previously established study findings of 15-20% of inflammatory etiology.
After starting the patient on treatment, monitoring of disease activity and response to treatment can be assessed by the following clinical and lab parameters.
CLINICAL
• Existing lesions of vasculitis starts healing which is made out clinically by fading out of maculo-papular dusky red erythematous lesion leaving a hemosiderin induced post-inflammatory hyperpigmentation.
• No new lesions of vasculitis should be encountered.
LAB PARAMETERS
• Fall in the titre of markers of acute inflammation like ESR & CRP.
• Fall in the titre of ANA, dsDNA and ANCA.
• Reduction in the Brimingham Vasculitis Activity Score (BVAS)
Patients have to be monitored with repeated blood tests, once in 3 months to watch for markers of further progression of disease in the form of appearance of neoantigens and antibody like dsDNA becoming positive later in the course of the disease.
In our study, in majority of patients (53%), the etiology was idiopathic. Vasculitis in the background of SLE contributed to 12 patients (16%). ANA titre and other antibodies were monitored more diligently in those patients.
In our study, one patient who was recalcitrant to treatment progressed to ANCA positivity during the course of illness and developed endophthalmitis and interstitial lung disease.
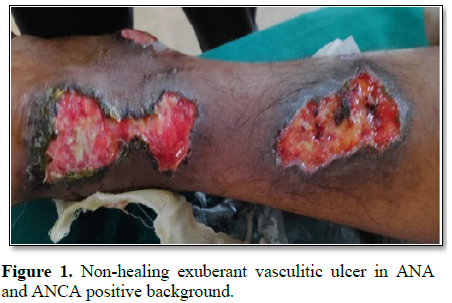
We also encountered a young female patient of age 25 years who presented with seizures and malignant hypertension with spontaneous non-healing ulceration in both legs. Her ANA and ANCA profile initially was negative and despite treatment, her ulcers didn’t heal. On regular follow up of the patient, her ANA and ANCA became positive within a span of 6 months and were subsequently treated with steroids for Polyarteritis Nodosa (Figure 1).
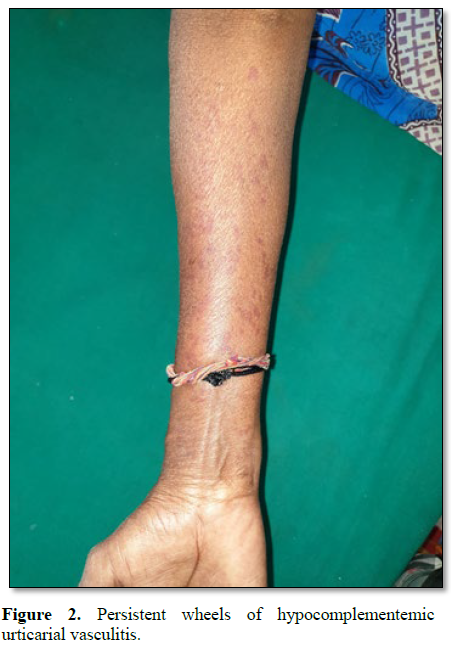
Similarly, a middle aged woman presented with recurrent episodes of persistent urticarial wheals over both extremities and trunk. Vasculitis workup was done. ANA was negative. CRP was found to be elevated (Figure 2).
As recurrent Urticarial Vasculitis may be a presenting feature of SLE and Sjogren’s syndrome [4], patients with this kind of presentation should be kept on a long term vigil, so as to detect evolving SLE and Sjogren’s earliest possible.
Earnest efforts should be made to reach a definite diagnosis in these types of patients, as majority of them have features suggestive of undifferentiated connective tissue disorders and overlap syndromes. This needs a comprehensive approach towards understanding of evolution of the disease process with respect to chronology of appearance of serological markers viz., varying Extractable Nuclear Antigens, the corresponding Auto-antibodies and the symptom complex resulting out of them. Predicting prognosis and choosing therapeutic options in these cases will by and large depend on the extent of organ involvement.
CONCLUSION
Benign Idiopathic CSVV is a self-limiting entity, settles down in 6 months to 1 year of treatment, provided patient does not develop any other surrogate markers like ANA, dsDNA and ANCA during the course of illness.
Patient of vasculitis who remains ANCA negative at the point of commencement of treatment, can become ANCA positive at any point of time, thereby signaling ominous sign, as with ANCA positivity, vasculitis starts involving medium sized and large sized vessels thereby leading to loss of function of vital organs, which could at times be fatal, as it can lead to conditions like Panophthalmitis, Endophthalmitis and Interstitial Lung Disease. If C-ANCA and P-ANCA coexist in a patient, drug-induced vasculitis should be suspected.
It was found out in our study that 80% of patients with CSVV responded well to systemic steroids, the simplest and safest immunosuppressant. Also it was inferred, that maintenance of patients with more than one immunosuppressant at a particular point of time is not advisable as the risk of complications like opportunistic infections, pancytopenias and avascular necrosis of femur, is on the higher side.
CSVV in ANA positivity background needs investigations and diligent follow up as it may develop into full blown SLE and hence patients have to be offered a guarded prognosis [5]. In tropical countries like India, Tuberculosis and SLE mimic each other closely in the early stages of evolution [6,7]. Hence sufficient precaution and exhaustive work up is always mandatory to rule out Tuberculosis before starting the patient on immunosuppressant.
1. Griffiths C, Barker J, Bleiker T, Chalmers B,
et al. (2016) Robert Rook’s Textbook of Dermatology. 9th Edn.
London, UK: Wiley Blackwell, pp: 1021-1028.
2. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid
MC, et al. (2013) 2012 revised International Chapel Hill Consensus Conference
Nomenclature of Vasculitides. Arthritis Rheum 65: 1-11.
3. Calabrese LH, Michel BA, Bloch DA, Arend WP,
Edworthy SM, et al. (1990) The American College of Rheumatology 1990 criteria
for the classification of hypersensitivity vasculitis. Arthritis Rheum 33:
1108-1113.
4. Garcia‐Porrua C, Gonzalez‐Gay MA (1999)
Comparative clinical and epidemiological study of hypersensitivity vasculitis
versus Henoch‐Schonlein purpura in adults. Semin Arthritis Rheum 28: 404-412.
5. Watts RA, Jolliffe VA, Grattan CE, Elliott J,
Elliott J, Lockwood M, et al. (1998) Cutaneous vasculitis in a defined
population - Clinical and epidemiological associations. J Rheumatol 25:
920-924.
6. Russell JP, Gibson LE (2006) Primary
cutaneous small vessel vasculitis: Approach to diagnosis and treatment. Int J
Dermatol 45: 3-13.
7. Lotti T, Ghersetich I, Comacchi C, Jorizzo JL
(1998) Cutaneous small-vessel vasculitis J Am Acad Dermatol 39: 667-687.
QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Carcinogenesis and Mutagenesis Research (ISSN: 2643-0541)
- Journal of Neurosurgery Imaging and Techniques (ISSN:2473-1943)
- Journal of Rheumatology Research (ISSN:2641-6999)
- Journal of Psychiatry and Psychology Research (ISSN:2640-6136)
- International Journal of Internal Medicine and Geriatrics (ISSN: 2689-7687)
- Chemotherapy Research Journal (ISSN:2642-0236)
- Advance Research on Alzheimers and Parkinsons Disease


