1212
Views & Citations212
Likes & Shares
Although much
effort has gone into genomic sequencing to define disease, the downstream
products of gene sequences-proteins, nevertheless remain the master regulators
of biology via their interactions with nucleic acids and other macromolecules.
Many proteins are measurable in blood, making it a rich resource for
biomarkers. Yet for reasons largely unrelated to analytical limitations, this
resource remains largely untapped. In this review, we describe how chronic
illness manifests itself in blood and how we might study innate immunity to
understand mechanisms that can potentially translate into new biomarkers and
therapeutic modalities. We draw upon our own knowledgebase of proteome
information reportable after using depletion or enrichment products in LC-MS/MS
workflows and how this knowledge can be utilized in new strategies for
biomarker discovery from blood samples. We note that BSG’s products have simply
and efficiently reduced the complexity of the serum proteome allowing for
cost-effective workflows, without the use of antibody-based depletion methods.
Finally, we discuss how patterns of Serpins, a superfamily of protease
inhibitors, may serve as a surrogate measure of the progressive stages of the
innate immune systems’ response
to both infectious and non-infectious disease. This
convergence of strategies and LC-MS/MS technologies has made the task
immediately available to investigators to now develop the next generation of
molecular tests for more precise and personalized treatment of patients.
INTRODUCTION
Blood is the body’s vehicle for the
accumulative evidence of pathological insults for diseases. Secreted proteins,
extracellular vesicles and circulating blood cells mediate individualized
homeostasis via intercellular communication, immune responses, vascular and
endothelial cell function, tissue remodelling, fluid exchange and nutrient assimilation
[1]. Thus, plasma/serum proteins and other circulating factors directly
regulate complex processes such as aging and the development of common chronic
diseases. Most diseases are multi-factorial with many proteins collectively
acting within highly regulated networks-even single gene diseases can be at the
centre of larger, complex regulatory networks.
As such, a disease state results whenever this
protein network becomes dysregulated over long periods via a confluence of
heredity, lifestyle, or environmental stimuli. Because blood mediates
coordination between nonadjacent tissues, it is essential to understand how
this dysregulation manifests itself, regardless of the underlying causative
factors. Quantitative proteomics from blood can help unravel these regulatory
elements. Yet, extracting and characterizing functional changes and adaptations
to disease for many of even the highest abundance proteins in circulation
remains limited. We therefore propose new strategies to support proteomic
analysis of blood.
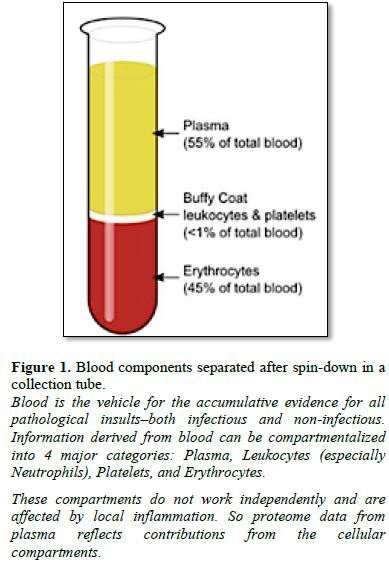
Proteins in blood can be separated into four main compartments-red cells, white cells, platelets and plasma (Figure 1). Serum differs from plasma primarily in the amount of Fibrinogen, however, for purposes of this review, the concepts proposed herein consider that proteome information derived from serum or plasma would be similar.
For that reason, the terms are used interchangeably
here.
In this review, we describe how
blood cells release inflammatory cargo proteins, disrupting the delicate
balance necessary for normal homeostasis. More precisely, information about the
function and communication between proteins and blood cells will ultimately be
the best possible information that can be derived to understand disease states
[1]. Consequently, proteomic biomarker panels from blood will become highly
beneficial, as blood is a relatively non-invasive sample type that can be
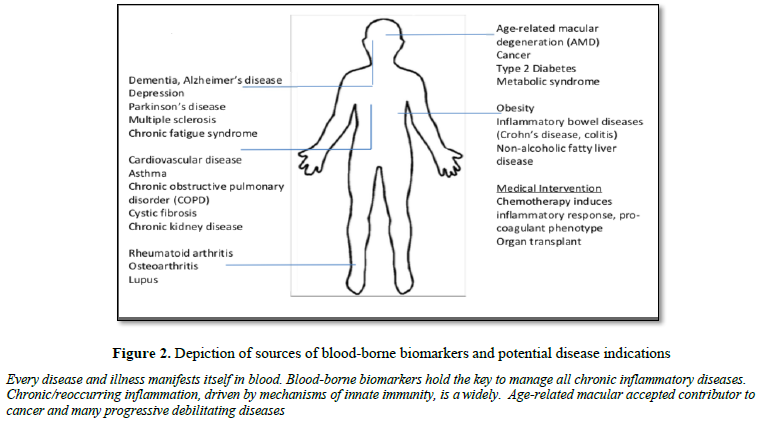
monitored longitudinally throughout a lifetime (Figure 2).
Blood proteomic analysis (e.g., by LC-MS/MS) does however, have inherent challenges. Serum proteomics can be especially challenging for two reasons: 1) the presence of highly abundant proteins, e.g., albumin alone accounts for about 50% of the total protein mass and 2) immunoglobulins, a proteolytically resistant protein family. Several sample preparation strategies are used to address these challenges, most of which employ the use of immuno-affinity depletion to remove one or more high abundance proteins. Common limitations of immuno-affinity however are high costs, regeneration requirements, which may result in a diminished and inconsistent performance and a required marriage of species to antibody. Because of these limitations, Biotech Support Group (BSG) has developed products and methods that are not based on immuno-affinity, but rather are derived from non-biological bead-based chemistries. These have proven advantageous in a variety of LC-MS/MS workflows [2-6].
With some
modest adjustments, Viaralet et al. [5] concluded that the BSG product -
AlbuVoid™ - proved to be faster and more cost-effective than antibody-based
methods to improve quantitative clinical proteomics. Furthermore, BSG’s
HemoVoid™ proved especially useful for annotating erythrocytes in the human
proteome project to identify additional proteins and N-termini; 778 proteins
were identified from the cytosolic fraction, 171 of which were not represented
in either the soluble non-depleted fraction or the membrane fraction [6]. Based
on BSG’s internal investigations, we have accumulated data that encompasses
greater than 1000 serum proteins that can be observed by LC-MS/MS, and
categorically characterized with respect to the proposed strategy described
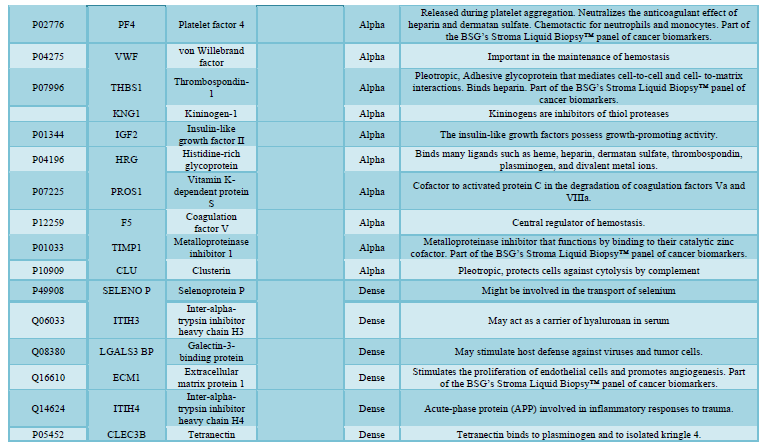
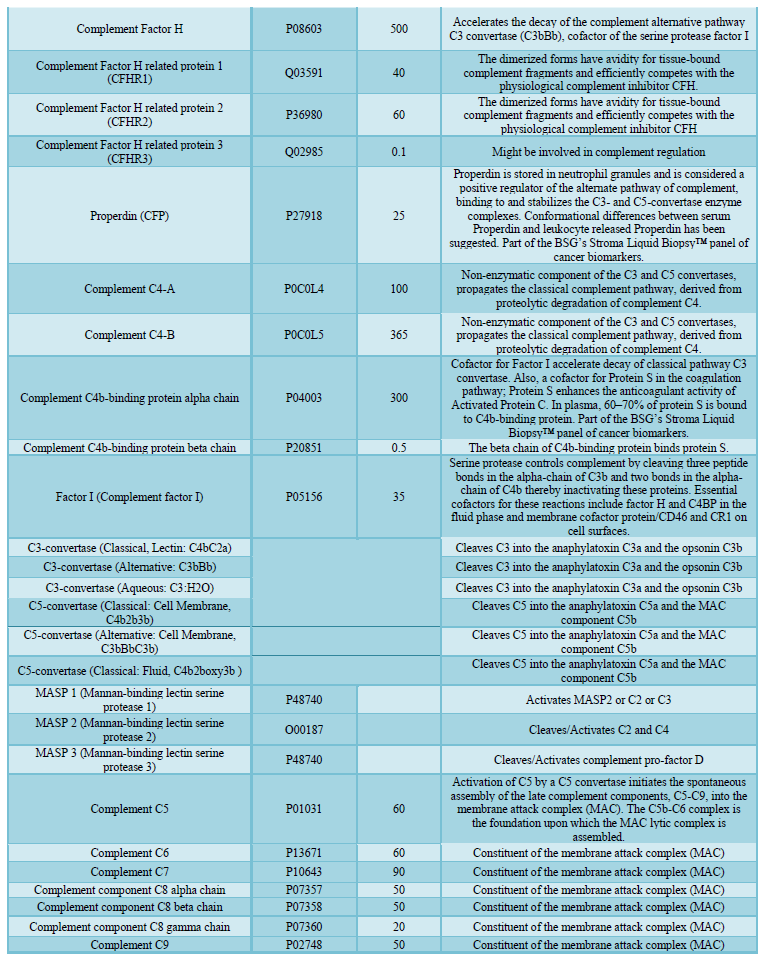
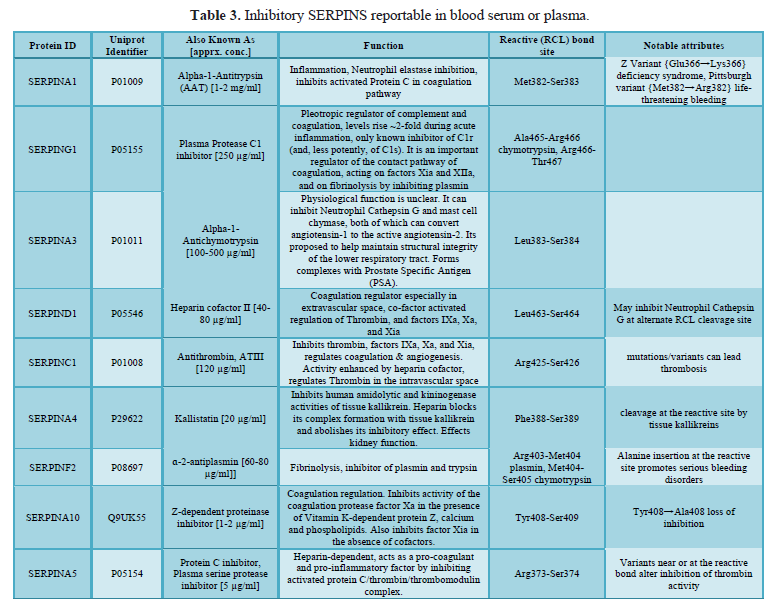
here (Tables 1, 2 and 3).
While acknowledging some exceptions, notably C-reactive protein (CRP) or
antibodies, consequential changes in the blood proteome are not necessarily
derived from tissue leakage, but rather from proteolytic modifications,
presumably driven in large part by inflammatory release of blood’s cellular
protein cargo. Within the coagulation/complement axis, many proteins circulate
as inactive precursors (zymogens) and
only become active upon proteolysis. Once activated, protein sub-forms then
become ligands for cell receptors, substrates for other proteases in cascading
sequences, or interacting partners for regulating mechanisms involving all
blood cells, vessel walls and vasculature, along with other plasma co-factors
(i.e., lipids, Heparins, metal cations, etc.). For these reasons, we examine
new ways to observe and measure such categorical and functional responses to
inflammatory disease and related disorders.
Unlike proteins from tissue, the vast majority
of proteins contained in blood, either cellular or humoral (extracellular), are
quite constant; blood’s cellular content being derived either from cells
without nuclei (red cells and platelets) or from those with lobed nuclei with
limited new protein production capacity (neutrophils). Thus, differential
changes are derived from a host’s systemic response to many varieties of
environmental stimuli (both infectious and non-infectious), not from altered
gene expression. As such, we make the case in this paper for proteomic analysis
of blood to become a discipline of categorical
classification and functional metrics, rather than a discipline of
finding the exceedingly low concentration (<
Furthermore, categorical metrics have the
advantage of monitoring proteins in a highly observable LC-MS/MS concentration
range, ≥ 0.1 µg/ml in most cases.
This new strategy, to represent proteome
information within categories, is designed to derive characteristic patterns,
rather than single biomarkers, that can support clinical manifestations of
disease or response to medical intervention. By categorical classification,
we hope to gain a much deeper understanding of the molecular relationships that
are shared amongst apparently distinct pathological phenotypes. This would help
explain, for example, the increased risk of cancer in patients with
inflammatory bowel disease, or the apparent link between Rheumatoid arthritis
or lupus with increased risk of blood clots. Finally, once we understand these
shared relationships, we hope to address therapies that may have been developed
for one clinical condition and apply them towards other clinical phenotypes,
with biomarkers that can help guide selection and utility.
While important, we have purposely left out the influence of the red cell
proteome. This is because there is limited information on the role of
erythrocytes, with notable exceptions (i.e., Paroxysmal nocturnal
haemoglobinuria), in the central theme of this review, namely, the
orchestration of the innate immune response.
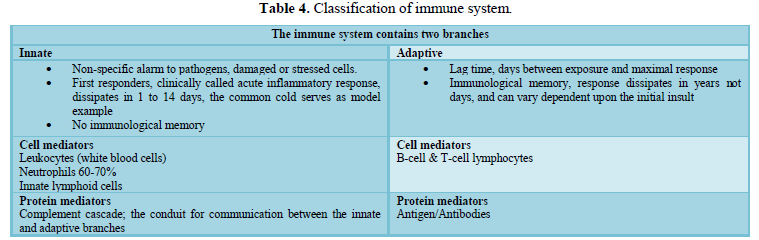
INNATE IMMUNITY
Innate immunity refers to first-line,
non-specific defense mechanisms that come into play immediately or within hours
of a perceived pathogenic insult in the body. The innate immune response
consists of physical, chemical and cellular defences against pathogens. Its
main purpose is to immediately prevent the spread and movement of foreign or
non-self-pathogens throughout the body and to initiate the second line of
defence, the adaptive or acquired immune response. As a second-line defense,
the adaptive response occurs downstream from the innate immune response and
starts transferring immunological longer term memory towards the
non-self-pathogens. So a normal resolution of the innate response leads to a productive
handoff to the adaptive response. Conversely, an unresolved innate response may
delay or confound a suitable adaptive response, with both acute and chronic
disease consequences.
Taken together, the human immune system has evolved to adapt and respond
to a variety of physical insults and never-ending exposure to infectious agents
to survive. While the emergence of COVID-19 reminds us that infectious insults
remain a large healthcare threat, mankind’s pharmacological skills (e.g.,
antibiotics, vaccines, etc.) has largely eliminated many infectious insults of
the past. As a result, we are able to live longer than our predecessors.
Unfortunately, our pharmacopeia and inflammatory response systems are not
sufficiently capable of fending off today’s longer-lifetime exposure to
environmental and lifestyle insults that we now face. These exposures and
insults contribute to chronic inflammation which, over time, are manifested in
a variety of pathological conditions, including an important link between the
coagulation and innate inflammatory system [7, 8].
Thrombo-inflammation describes a process by
which the activation of coagulation assists the function of the innate immune
system and, conversely, components of the immune system contribute to
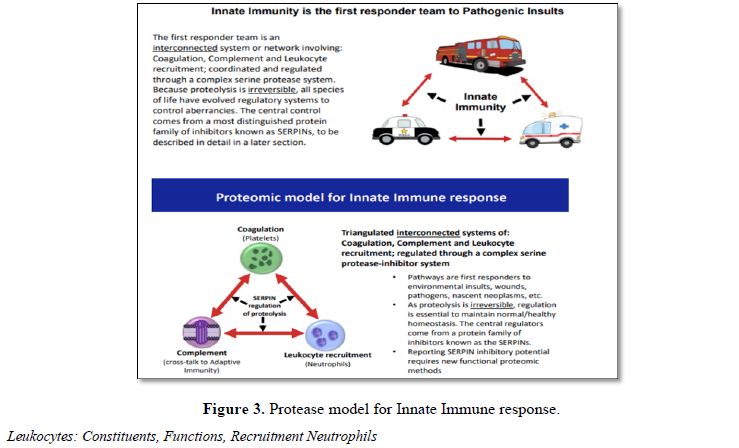
thrombosis. Thus, the innate immune system is an integrated triangulated
network that when functioning properly provides steady state control of
pathways involved with coagulation, complement and leukocyte recruitment (Table
4 and Figure 3). Any dysregulation within one affects the others and so
collectively, a dysregulated innate immune system can contribute to the genesis
of many acute and chronic pathologies. For example, chronic inflammation is
widely recognized as a potential contributor to cancer and many progressive
debilitating diseases (e.g., inflammation due to H.pylori leads to stomach
lesions and cancer). As witnessed with COVID-19, the pre-existing inflammatory
status, may be a key factor to the acute severity of disease upon exposure to
infectious pathogens.
Because the innate immune system does not
change genetically over time, measuring panels of its protein level
orchestration is a worthwhile goal of proteomic investigation. Because of the
many feedback signals necessary to maintain steady state within the network,
therapeutically modulating even one rogue component may ultimately unwind the
overall dysregulation and contribute to longer term management of the disease.
For these reasons, we have focused this review on key regulatory elements of
innate immunity that can be observed, reported and surveyed by proteomic
analysis of serum/plasma. Within this model system, we describe protein
contributions from each pathway and new strategies for proteomic categorization,
function and disease characterization.
White blood cells (WBCs, also called leukocytes) are the cells of the
immune system that are rapidly recruited to protecting the body against both
infectious disease and perceived environmental insults. When anti-coagulated
whole blood is centrifuged in collection tubes, the white blood cells (WBCs)
form a thin, white layer of cells (the buffy
coat;) between the sedimented red blood cells (erythrocytes) and plasma
(see tube in Figure 1). WBCs make up approximately 1% of the total blood
volume in a healthy adult, substantially less numerous than the red blood cells
(about 45%). Having
nuclei distinguishes WBCs from the enucleated red blood cells (RBCs) and
platelets, yet, even with nuclei, most are terminally differentiated and do not
undergo cell division in the bloodstream.
White blood cells are composed of differentiated constituents, each having
specific functions. Neutrophils are the most abundant white blood cell,
constituting 60-70% of the circulating leukocytes and they thus contribute to the observable blood proteome much more
so than the rest of the white blood cell constituents
combined. Neutrophils have extensive crosstalk with each of the major
blood cell subsets (megakaryocyte/platelets, myeloid and lymphoid) and other
extracellular soluble proteins contained in plasma, further magnifying their
importance in health and disease.
For example, changes in circulating leukocytes
including the ratios of neutrophils-to-lymphocytes (NLR),
monocytes-to-lymphocytes (MLR) and platelets-to-lymphocytes (PLR), have been
used to predict tumor occurrence and prognosis [10]. During the beginning
(acute) phase of inflammation, neutrophils migrate to the site of inflammation,
particularly as a result of infection, environmental stimuli, or onset of
cancer. Inflammation follows chemotactic signals such as Interleukin-8 (IL-8),
C5a from Complement activation, as well as other peptides and small molecules.
Neutrophils, as “first responders” release a variety of proteases (i.e.,
Elastase) to remodel the extracellular matrix of the tissues to which they
migrate.
Once arrived, neutrophils then release granule
proteins and/or chromatin (DNA & histones) to form an extracellular fibril
matrix known as Neutrophil extracellular traps (NETs). Such NET release has
been observed to occur not only during acute (bacterial or viral) inflammation but
also in numerous pathological conditions, such as autoimmune diseases, vascular
diseases, and cancer [11]. In addition to NETs, neutrophil granules can be
secreted extracellularly (released cargo) in response to localized
immunological stimuli. As neutrophils have limited capacity for de novo protein
synthesis, this released cargo thus becomes a subset proteome that collectively
reflects a weighted average of cell maturities and the inflammatory stimulus
that triggered the release. These released proteins can be observed and
reported in the general blood circulation (many in our BSG serum knowledgebase,
see Table 1).
Platelets are rapidly deployed
to sites of injury or infection, and can modulate inflammatory processes by
interacting with neutrophils, forming platelet-neutrophil aggregates. Platelets
play a central role in innate immunity, initiating and participating in
multiple inflammatory processes, operating in parallel with their better known
clotting function. Platelets contain dense granules, alpha granules and
cytosolic proteins; granule secretion being pivotal to establishing and
controlling the microenvironment at the local inflammatory site.
Granule cargo release is both
contextual and kinetically controlled, mediating early activation, or
persisting long after the initial stimuli. Persistence of Platelet-released
along with Neutrophil-released cargo in unresolved chronic inflammation,
necessitates a systemic response to regulate their effects, proteolysis being
the most deleterious [12].
Granule
characteristics
1. α granules (alpha granules) – contents
include insulin-like growth factor 1, transforming growth factor beta (TGF-β),
platelet factor 4, and other clotting proteins (such as thrombospondin,
fibronectin, factor V, and von Willebrand factor) [13].
2. δ granules (delta or dense granules) –
contents include adenosine di- & tri-phosphates, proteins, and the majority
of platelet calcium, an essential co-factor in the coagulation cascade [13].
Like neutrophil granule cargo, this released cargo thus becomes a subset
proteome that collectively reflects the inflammatory stimulus that triggered
the release. Released protein cargo from these different granules thus forms a
signature characteristic of inflammatory responses that can be observed in
serum and reported (Table 1). Differences in quantitative elements from
one granule vs. another may be informative as to the relative weight that one
granule type cargo has to a particular disease phenotype. The released cargos
from both neutrophils and platelets simultaneously imparts their net effects on
the third component of the triangulated network – the complement cascade.
COMPLEMENT CASCADE
The complement cascade is a major component of
the immune system that provides powerful host surveillance and protection from
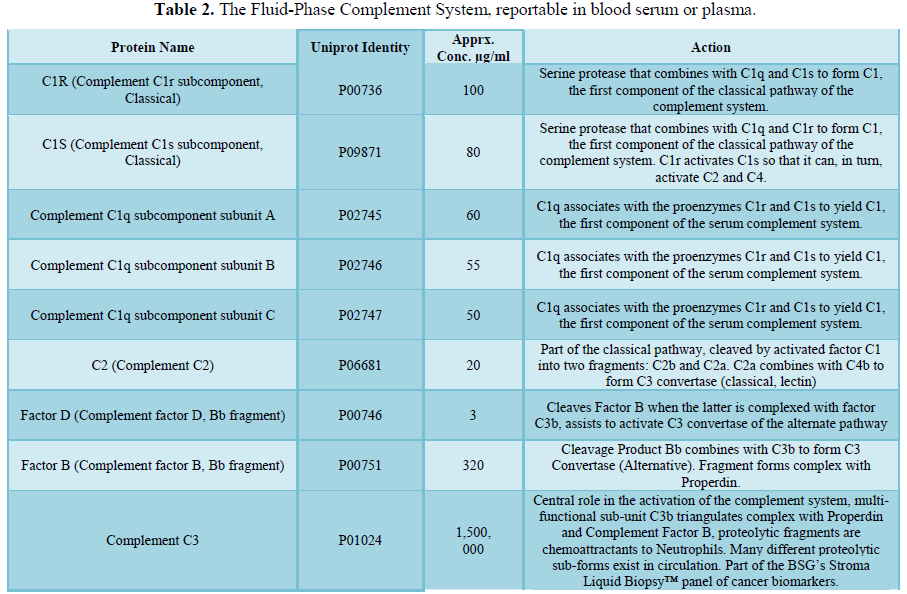
invading microbes. Comprising about 5% of the total protein mass in plasma (Figure
1 and Table 2), most complement proteins circulate in blood as inactive
precursors (zymogens); when triggered, these zymogens become activated through
proteolytic cascades. These cascades enhance the ability of antibodies and
phagocytic cells to clear microbes and damaged cells as well as promote local
inflammation.
The complement system links the innate immune
system to the adaptive immune system. This is a critical juncture; a delicate
balance must be maintained to allow activation when necessary to counteract
foreign or modified self/host surfaces, while concurrently protecting healthy
self/host tissue [14]. Protection is achieved systemically through the
concerted action of activators and inhibitors - about 50 membrane-bound and
soluble proteins - that ensure cell and tissue integrity essential for normal
health and well-being. When dysregulated, pathological conditions can arise,
including neurodegenerative diseases, cancer, age-related macular degeneration
(AMD), membranoproliferative glomerulonephritis, systemic lupus erythematosus,
transplant rejection, paroxysmal nocturnal haemoglobinuria (PNH),
ischemia-related conditions and autoimmune disease, to name a few [14,15].
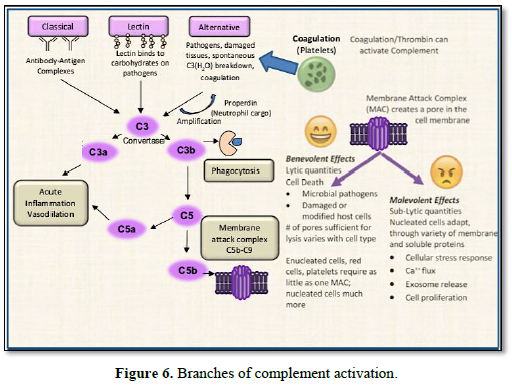
The three best characterized branches of
complement activation are: the classical, lectin, and alternative pathways, as
shown in Figure 6.
1. The classical pathway is initiated
by the C1 complex binding to multimeric antibody complexes, leading to cleavage
of C4 and C2 components and formation of the classical C3 convertase, C4bC2a.
2. The lectin pathway is activated by
binding of mannan-binding lectin (MBL), or ficolins (secreted, lectin- type
pattern recognition receptors) to carbohydrate or acetylated groups on target
surfaces. MBL and ficolins interact with MBL-associated serine proteases (MASP)
leading to cleavage of C4 and C2 and formation of the classical C3 convertase,
C4bC2a [16]. Noteworthy is that one of the neutrophil granules previously
described, is Ficolin-rich; another
example of the many network associations between cellular and extracellular
events occurring within the innate immune response.
3. Alternative pathway activation
involves interaction of C3(H2O) and/or previously generated C3b with
factor B, which when cleaved by factor D, generates the alternative C3
convertases C3(H2O)Bb and/or C3bBb. Complement related proteins are
always present in the blood and a small percentage spontaneously activate (Table
1). At about 1.5 mg/ml concentration in serum, C3 is by far the most
abundant. Notably, low-level hydrolysis of the C3b thioester bond - also known
as the C3 ‘tick-over’ notated as C3 (H2O) - keeps the alternative
pathway in constant alert to pathogen challenge.
All three pathways converge upon generation of the C3 convertase
complexes, facilitating the proteolytic cleavage of C3 to generate split forms
opsonin C3b and anaphylatoxin C3a. C3b covalently binds to proteins distributed
across the target cell surface. This is followed by an amplification reaction
that generates additional C3 convertases and deposits more C3b at the local
site. C3b can also bind to C3 convertases switching them to C5 convertases,
which then cleaves C5. Thus, by whichever initial means of activation, the
complement cascade system leads to one or more important final outcomes:
1. Opsonization of pathogens or damaged-self cells to enhance
phagocytosis,
2. Production of anaphylatoxins C3a & C5a involved in the acute
inflammatory response,
3. Recruitment of leukocytes to the inflammatory site,
4. B- and T-cell stimulation, and
5. The terminating end of the cascade – assembly of the membrane attack
complex (MAC) on the cell surface.
The terminating end of the complement cascade
is derived from the C3 Convertase proteolytic product - C5b, which engages the
sequential recruitment of C6, C7, C8, and C9, assembling the MAC. Also known as
the “terminal complement complex”, it results from the coordination of C5b-7
insertion in the membrane, which then captures C8, inducing polymerization of a
C9 ring – to as many as 18, C9’s per pore. Terminal MACs
punch a hole (pore) in the membrane of the invading pathogen or target
cell, and when a sufficient number of MAC pores form, the cell dies by osmotic
lysis.
However, when insufficient quantities of MAC or when sufficient
inhibitors of MAC are present, sub-lytic conditions persist that are
non-lethal, and unable to kill the cell. Instead, these conditions promote
other cellular stress adaptations thereby setting up the duality of outcomes
imposed by Complement activation. At sub-lytic doses, the complement MAC has
wide ranging effects that differ based
on the cell types, leading to a variety of
cellular responses, such as extracellular vesicle secretion, aggregation and
chemotaxis. Sub-lytic Complement can also induce increased cell resistance to
lytic doses of Complement [17]. These can all conspire to have pathogenic
consequences. Complement regulators expressed at high levels on malignant cell
membranes form an escape mechanism from MAC, setting up sub-lytic
concentrations that can activate intracellular signals leading to malignant
cell proliferation [18, 19].
While traditionally described as three
activation pathways, an additional, largely under-appreciated pathway is
Complement’s evolutionarily conserved link to coagulation to eliminate damaged
tissues [20]. C1 Inhibitor (Serpin G1; see next section for further details)
serves as one model protein mediator of this linkage, as it acts to inhibit
both complement activation and coagulation initiation. As another example,
C4b-binding protein is a cofactor for both Factor I (a Complement regulator)
and Protein S (a Coagulation regulator). Also, Complement may activate
platelets or facilitate biochemical and morphological changes in the vascular
endothelium, thus potentiating coagulation and contributing to haemostasis in response
to injury [21, 22]. Conversely, Complement can be activated from proteolytic
enzymes derived from coagulation and fibrinolysis that can cleave both C3 and
C5 [23, 24]. Consequently, the Complement system adapts to, and is influenced
by many different proteolytic triggering events.
Such events can also be influenced by
competing factors from coagulation and neutrophil recruitment. For example, in
mouse models of fatal injury deprived of neutrophils, amplification of
complement can occur, which suggests infiltrating inflammatory cells
participate in localized Complement activation [25]. Likewise, platelets
adhered to injured vessel walls form strong adhesive substrates for leukocytes,
a major event in thrombo-inflammation. This series of responses is mediated by
Complement and specifically to alternative Complement activator Properdin,
derived in part from neutrophil granule cargo [8].
It has also been proposed that the release of Properdin from neutrophils
is both a major determinant of local Complement activity and behaves
differently from endogenous serum Properdin [26]. In like manner, Properdin
released from neutrophils, is structurally related to Thrombospondin-1 (THBS1),
a protein released from platelet cargo, both containing conformationally diverse
Thrombospondin Type 1 Repeats (TSRs) [27,28]. It is interesting to speculate
how TSR binding site competition might influence Complement regulation (one
example being THBS1 interaction with Alternative Complement regulator-Factor H
[29]). Significantly, several reports suggest Complement-mediated interactions
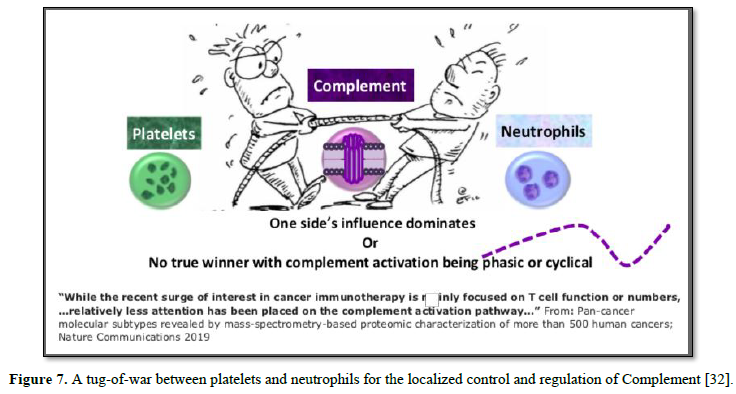
between Neutrophils and Platelets [30, 31]. Regardless of the underlying
mechanism, the end result may well be a tug-of-war between platelets and
neutrophils for the localized control and regulation of Complement [32] (Figure
7).
These
triangulated pathways cooperatively form an intensified cycle resulting in
local inflammation, thrombosis, and tissue damage. Chen et al. [33] illustrates
how this all may play out in disease. In that study, proteomic analysis
classified tumor tissues of over 500 cancers into just 10 subtypes, regardless
of the primary tumor of origin. Out of the 10, four subtypes were based solely
from the host’s stromal character at the local microenvironment, unrelated to
cellular mutations. Of the four stromal subtypes, two reflected immune
components: the first by presence of immune T cells, while the second immune
subtype, was highlighted by complement activation [32].
Taken together, these studies illustrate how the complement system orchestrates host defense by sensing a danger signal, translating the signal into specific cellular responses, and concurrently, communicating with other biological pathways ranging from adaptive immunity to hemostasis [34]. Such importance notwithstanding, there are limited tests available to assess these conditions in clinical practice. C3 and C4 are most frequently measured; total complement activity (CH50 lytic assay) can be measured if a deficiency is suspected. These tests, however, are non-specific and none can monitor sub-lytic activity, which can often be the most malevolent outcome of complement activation. Clearly, a better characterization of the total picture of Complement activity is needed. Proteomics can serve that need as it can identify and develop biomarkers to capture and report virtually all potentially competing influences, context dependencies and duality of outcomes imposed by Complement. In this regard, we propose here the functional profiling of the protease inhibitor family of Serpins as an important way to address this unmet need.
SERPIN Function and Regulation
Motivation
As proteolysis is irreversible, there is an
essential balance and regulation of proteolytic cascades necessary to keep
aberrancies controlled. Central to this maintenance are the Serpins. This
unique family of protease inhibitors serves the central control function of
innate immunity, in some way regulating all of its proteolytic mechanisms.
Our goal is to show how patterns of Serpins, a
superfamily of protease inhibitors, may serve as a surrogate measure of the
progressive stages of the innate immune systems’ response to a disease.
BACKGROUND
Serpins are a superfamily of proteins with
similar structures found in all kingdoms of life. They were first identified
for their protease inhibition activity (proteolysis regulation). Although some
proteins with Serpin sequence annotation are not protease inhibitors, but
instead perform diverse functions such as hormone transport, we focus in this
section on only serum Serpins with inhibitory activity.
Serpins are an extensively studied family of
both intercellular and extracellular proteins that exhibit conformational
adaptabilities and a most unique functional mechanism. Often called suicidal
inhibitors, inhibitory Serpins are in sharp contrast to the more conventional
competitive mechanism for protease inhibitors that bind to and block access to
the protease active site in a concentration dependent (Michaelis-Menten type)
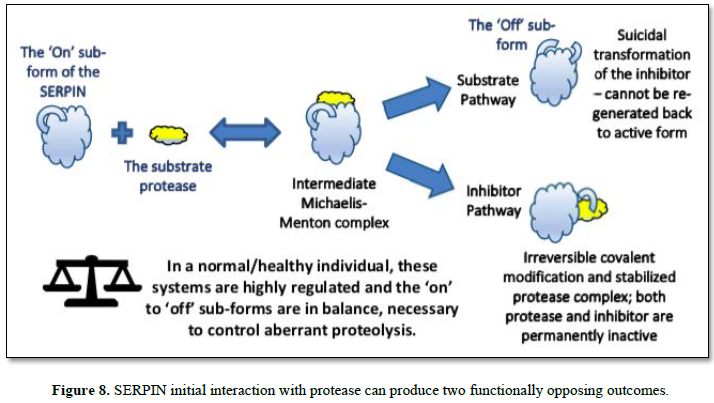
equilibrium. Such is not the case with Serpins, as the initial interaction
starts through a decoy process (an intermediate Michaelis complex). Here, the
protease sensing the Serpin as a suitable substrate, initially binds to a
peptide region of the Serpin – known as the reactive center loop (RCL) -that
transiently protrudes from the core of the Serpin body. From this intermediate
complex, one of two possible final outcomes are produced, called, respectively,
the Substrate and Inhibitor Pathways, as depicted in Figure 8, and
summarized as: follows:
In the inhibitor pathway, the protease
selectively cleaves a peptide bond in the reactive center loop (RCL) of the
serpin. The resulting acyl-enzyme undergoes an extensive and rapid
conformational change, with a 180 degree translocation of the attached
protease. During this process the active site is deformed, and hydrolysis
cannot be completed. This process results in an irreversible covalent complex.
Trapped in this suicidal covalent embrace, both the protease and the Serpin are
irreversibly modified and cannot be regenerated back to their active forms.
In the Substrate Pathway however, the protease
releases from the complex and remains active (the ‘On’ sub-form), but not so
for the inhibitor as the RCL region is cleaved and the inhibitor becomes
permanently inactive (the ‘Off’ sub-form) [35, 36].
Nine inhibitory Serpins account for ≥ 5% of the protein mass contained in serum (Table 4, see also top fraction in Figure 1). With such a large regulating influence in blood, Serpins are themselves subject to many co-factors (e.g., Heparin) that act upon their inhibition efficiency. Any aberrations in this finely tuned mechanism can lead to progressive loss of functionally active forms or the accumulation of inactive forms, Figures 8 & 9. Insufficient Serpin control of irreversible and inflammatory proteolytic activity can lead to systemic dysregulation of innate immunity and progression of disease.
Further confounding the significance of this bifurcated inhibition
mechanism is that measurements of Serpins have conventionally relied on
immunological (ELISA)-type assays that count all sub-forms in aggregate and
thereby assume homogeneous populations. This can lead to erroneous conclusions
about their functional role in disease. Fortuitously, the different active vs.
inactive Serpin sub-forms are reportable from biofluids using LC-MS/MS analysis
[37]. In our own analysis for cancer, we have identified two critical nodes in
this network relating to the progressive loss of functionally-active Serpins:
(A1) Alpha-1-Antitrypsin and (D1) Heparin Cofactor II [38]. These proteins are
included as part of Biotech Support Group’s Stroma Liquid Biopsy™ [39] panel of
cancer biomarkers.
Other reports corroborate these results that
tumorigenesis can systemically be characterized by chronic exhaustion of
inhibitory active Serpins and resulting increased protease activity (Figure
10).
Context Dependent Proteomic Categorization of Disease
In the course of this review, we have provided
a brief description of the three key innate immunity pathways, how they
interact with each other, and how they are all regulated through protease
activation balanced by protease inhibition, most notably through the Serpin
family. As noted in our review, these immune responses are extremely
context-dependent, e.g., the vasculature at inflammatory sites differs
significantly from the surrounding tissue. Many factors contribute to create
the context in such environments: platelets attached to vessel walls,
vasodilation, shear sensing receptors and the relative amounts of diffusive to
axial transport of solutes. Thus, the activation of cells and the release of
their cargo proteins at the local site offers the means for a categorical
deconstruction of the inflammatory elements of disease. Potential markers may
now be reported from blood through the protein cargos released from neutrophils
and platelets; these cargo proteins are readily observable through quantitative
LC-MS/MS analysis. We highlight these proteins in the BSG knowledgebase in Table
2.
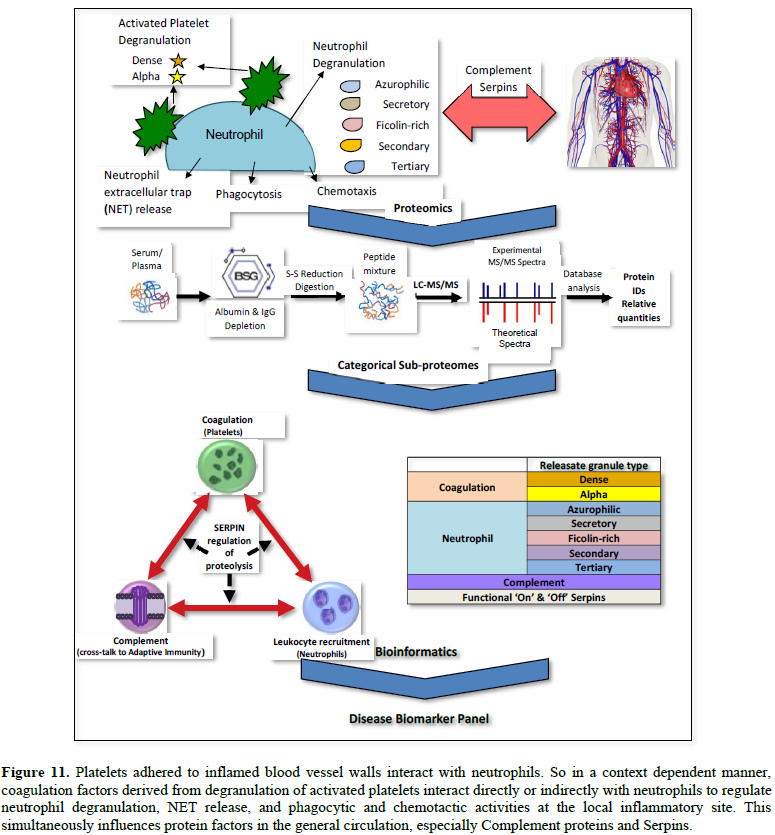
Finally, this context-specific, released
cargo enters the general circulation and simultaneously influences another tier
of response, namely, the highly observable and measurable Complement cascade
and Serpin regulation (Tables 3 and 4). All these various, systemic
responses can be monitored and categorized by quantitative proteomic analysis:
Platelet-released cargo, Neutrophil-released cargo, complement components and
Functional ‘On’ & ‘Off’ sub-forms of Serpins (Figure 11).
Through this new strategy, we posit that
biomarker panels can be derived from these categorical proteins which increase
or decrease in characteristic patterns from pathological conditions. In turn,
these panels can then be used to assess patients for actionable decisions.
This ground truth protein data creates a
survey of the function and communication between plasma and cells in blood.
With advancements in proteomics and bioinformatics for chronic diseases, we can
aspire to bring blood-based protein biomarkers to the clinic. Unlike single
markers such as HbA1c for diabetes management, future advances in protein
biomarkers will not come from one protein assay or even a small set of protein
assays, but from patterns of protein changes. The good news is that these
potential protein patterns are highly observable using LC-MS/MS and this
capability has become even more precise and comprehensive with improvements in
instrument speed and resolution. Advancements in the quantification of multiple
proteins at once, in both targeted label and label-free analysis; coincide with the use of internal standards and simple efficient sample prep workflows. This convergence of technologies has made the
task immediately available to those who, like us, seek to develop the next
generation of molecular tests for more precise and personalized treatment of patients.
1. M
Stastna, Eyk JEV (2012) Secreted proteins as a fundamental source for biomarker
discovery. Proteomics 12: 722-735.
2. Zheng,
H (2015) AlbuVoid™ coupled to on-bead digestion-tackling the challenges of
serum proteomics. J Proteom Bioinformatics 8:
225.
3. Biotech
Support Group Application Report (2019) AlbuVoid™ PLUS & AlbuSorb™ PLUS -
Evaluating Different Windows of
Observation Solves The Many Challenges of Serum Proteomics. Available online
at: https://www.biotechsupportgroup.com/v/vspfiles/templates/257/pdf/PLUS%20Application%20Report%2007212019%20v1.pdf
4. Klatt S, Roberts A, Lothian A (2020)
Optimizing red blood cell protein extraction for biomarker quantitation with mass spectrometry. Anal Bioanal Chem. Available online at: https://doi.org/10.1007/s00216-020-02439-5
5. Vialaret,
Jerome & Kadi, Sarah & Tiers, Laurent & O Flynn, Robin &
Lehmann, et al. (2018) Albumin depletion of human serum to improve quantitative
clinical proteomics. Curr Top Pept Protein
Res 19: 53-62.
6. Lange, Philipp F (2014) Annotating N termini for the human proteome project:
N termini and Nα-acetylation
status differentiate stable cleaved protein species from degradation remnants
in the human erythrocyte proteome. J Proteome Res 13: 2028-2044.
7. Esmon CT (2004) Interactions between the innate immune and blood coagulation systems. Trends Immunol 25: 536-542.
8. Swystun,
Laura L, Liaw PC (2016) The role of leukocytes in thrombosis. Am Soc Hematol
128: 753-762.
9. Blausen.com
staff (2014) Medical gallery of Blausen Medical 2014. WikiJournal of Medicine
1.
10. Feng,
Zhao S (2019) Elevated plasma fibrinogen and the neutrophil-to-lymphocyte ratio
as predictive risk factors for
prostate cancer. Int J Clin Exp Med 12: 13117-13126.
11. Kolaczkowska
E, Kubes P (2013) Neutrophil recruitment and function in health and
inflammation. Nat Rev Immunol 13: 159-175.
12. El
Rayes, Tina (2015) Lung inflammation promotes metastasis through neutrophil
protease-mediated degradation of Tsp-1. Proc Natl Acad Sci 112: 16000-16005.
13.
Reactome pathway
database (2019) Available online at: https://reactome.org/PathwayBrowser/#/R-HSA-76005&SEL=R-HSA-481033&PATH=R-HSA-109582,R-HSA-76002&FLG=P02776
14. Zipfel,
Peter F, Skerka C (2009) Complement regulators and inhibitory proteins. Nat Rev Immunol 9: 729.
15. Varela,
Carlos J, Tomlinson S (2015) Complement: An overview for the clinician. Hematology/Oncology Clinics
29: 409-427.
16. Ricklin,
Daniel (2010) Complement: A key system for immune surveillance and homeostasis.
Nat Immunol 11: 785.
17. Bohana-Kashtan,
Osnat (2004) Cell signals transduced by complement. Mol Immunol 41: 583-597.
18. Roumenina,
Lubka T (2019) Context-dependent roles of complement in cancer. Nat Rev Cancer 19: 1-18.
19. Afshar-Kharghan,
Vahid (2017) The role of the complement system in cancer. J Clin 127: 780-789.
20. Dzik,
Jolanta M (2010) The ancestry and cumulative evolution of immune reactions.
Acta Biochimica Polonica 57: 443-466.
21. Oikonomopoulou,
Katerina, et al. (2012) Interactions between coagulation and complement-their
role in inflammation. Springer-Verlag, 2012.
22. Markiewski
MM, B Nilsson, KN Ekdahl, TE Mollnes, JD Lambris (2007) Complement and
coagulation: Strangers or partners in crime? Trends Immunol 28: 184-192.
23. Amara,
Umme (2010) Molecular intercommunication between the complement and coagulation systems. J Immunol 185: 0903678.
24. Krisinger
MJ, Goebeler V, Lu Z, Meixner SC, Myles T, et al. (2012) Thrombin generates
previously unidentified C5 products that support the terminal complement
activation pathway. Blood 120: 1717-1725.
25. Girardi,
Guillermina (2003) Complement C5a receptors and neutrophils mediate fatal
injury in the antiphospholipid
syndrome. J Clin Invest 112: 1644-1654.
26. Kemper,
Claudia, Hourcade DE (2008) Properdin: New roles in pattern recognition and target clearance. Mol Immunol 45: 4048-4056.
27. Cortes,
Claudio (2013) Local release of properdin in the cellular microenvironment: Role
in pattern recognition and
amplification of the alternative pathway of complement. Front Immunol 3: 412.
28. Crombie,
René (2000) Mechanism of thrombospondin-1 anti-HIV-1 activity. AIDS Patient
Care STDs 14: 211-214.
29. Resovi,
Andrea (2014) Current understanding of the thrombospondin-1 interactome. Matrix
Biology 37: 83-91.
30. Blatt,
Adam Z, Pathan S, Ferreira VP (2016) Properdin: A tightly regulated critical inflammatory modulator. Immunol Rev 274: 172-190.
31. Saggu,
Gurpanna (2013) Identification of a novel mode of complement activation on
stimulated platelets mediated by
properdin and C3 (H2O). J Immunol 190: 6457-6467.
32. Chen,
Fengju (2019) Pan-cancer molecular subtypes revealed by mass-spectrometry-based proteomic characterization of more than
500 human cancers. Nat Commun 10: 1-15.
33. Merle,
Nicolas S (2015) Complement system part I-molecular mechanisms of activation and regulation. Front Immunol 6: 262.
34. Nording,
Henry, Langer HF(2018) Complement links platelets to innate immunity. Semin
Immunol 37.
35. Law,
Ruby HP (2006) An overview of the serpin superfamily. Genome Biology 7: 216.
36. Khan,
Sazzad M (2011) Serpin inhibition mechanism: A delicate balance between native metastable state and polymerization. J Amino
Acids 2011.
37. Roy,
Swapan, Kuruc M (2019) Methods to Monitor the Functional Subproteomes of SERPIN Protease Inhibitors. Functional Proteomics. Humana Press, New
York, NY, pp: 41-54.
38. Verhamme,
Ingrid M (2019) Loss of Functional Alpha-1-Antitrypsin and Heparin Cofactor II
in Inflammation and Cancer. The Serpins 2019 Conference, September 19-22, 2019
in Sevilla, Spain.
39. Stroma
Liquid Biopsy™ (2019) Blood-based biomarkers to monitor stromal conditioning in
cancer. Whitepaper
40. Sun,
Zhifu, Yang P (2004) Role of imbalance between neutrophil elastase and alpha
1-antitrypsin in cancer development
and progression. The Lancet Oncology 5: 182-190.
41. Janciauskiene,
Sabina (2001) Conformational properties of serine proteinase inhibitors
(serpins) confer multiple pathophysiological roles. Biochim Biophys Acta 1535: 221-235.
42. Manco-Johnson
MJ (1994) Antithrombin III. Anticoagulant: Physiologic, pathologic, and pharmacologic. CRC Press, Boca
Raton, Florida, USA Chapter 3: 27-40.
43. Hatto,
MW, Hoogendoorn H, Southward SM, Ross B, Blajchman MA (1997) Comparative
metabolism and distribution of rabbit heparin cofactor II and rabbit
antithrombin in rabbits. Am J Physiol 272: E824-E831.
-
 Table 1
Table 1 -
Table 2
-
Table 3
-
Table 4
-
Table 5
-
Table 6
-
Table 7






QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Genetics and Cell Biology (ISSN:2639-3360)
- Journal of Veterinary and Marine Sciences (ISSN: 2689-7830)
- Journal of Astronomy and Space Research
- Journal of Biochemistry and Molecular Medicine (ISSN:2641-6948)
- Journal of Genomic Medicine and Pharmacogenomics (ISSN:2474-4670)
- Journal of Agriculture and Forest Meteorology Research (ISSN:2642-0449)
- Journal of Womens Health and Safety Research (ISSN:2577-1388)


