4159
Views & Citations3159
Likes & Shares
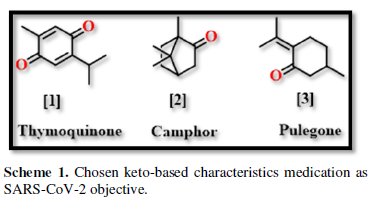
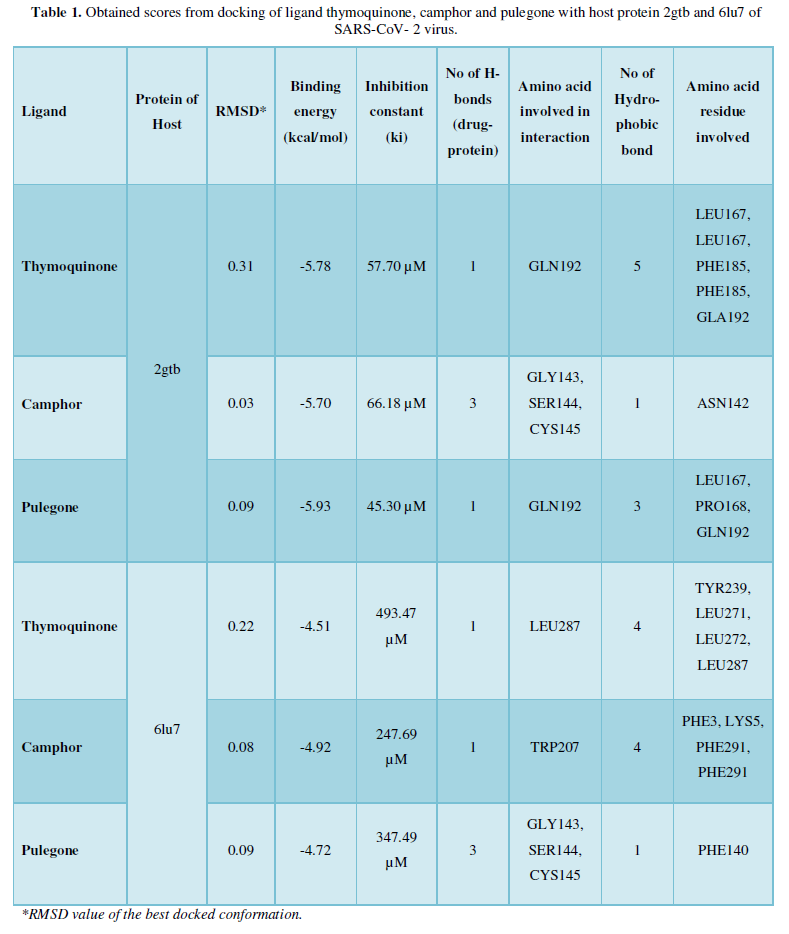
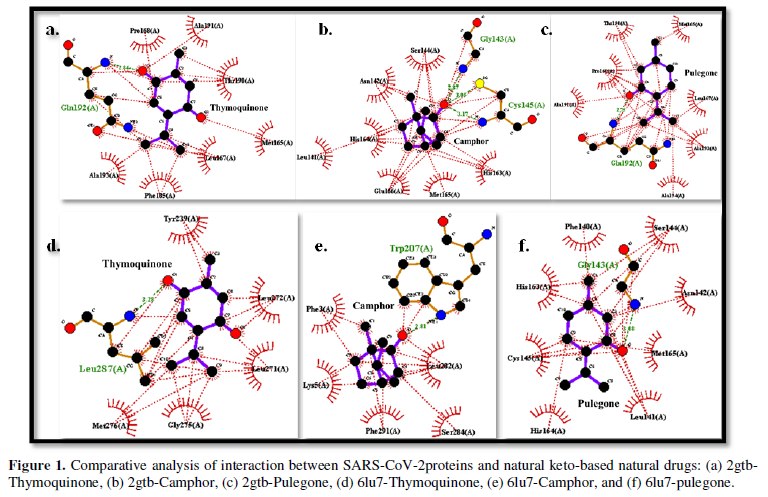
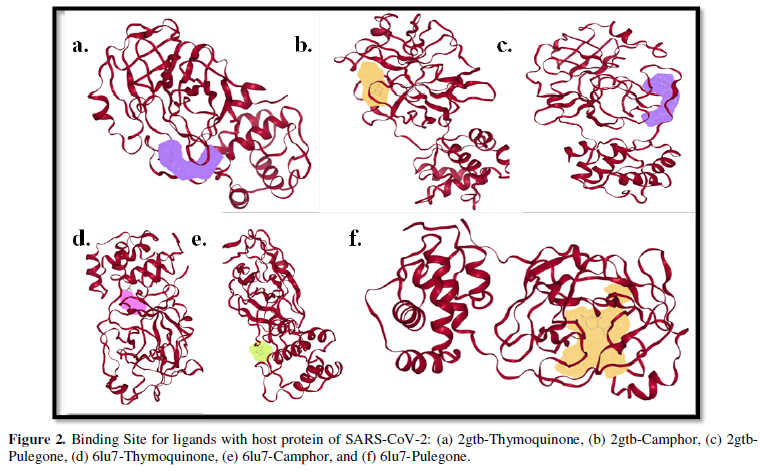
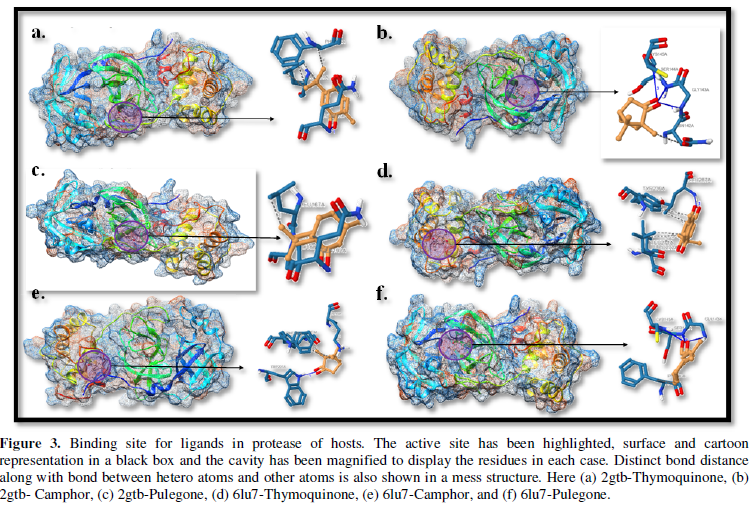
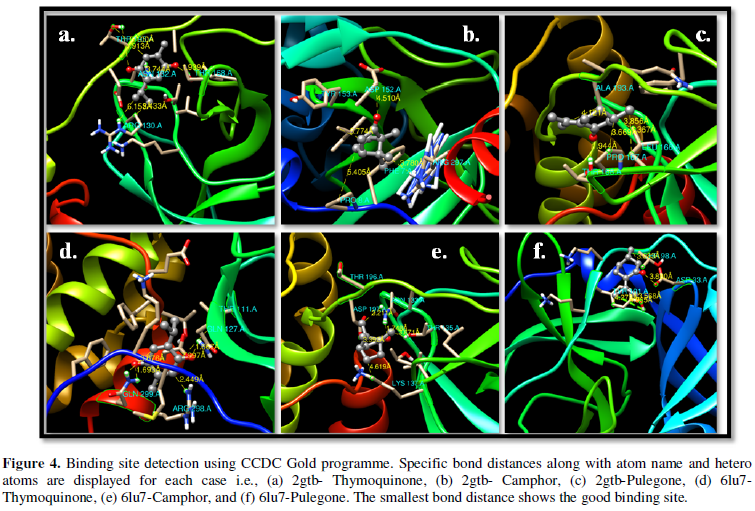
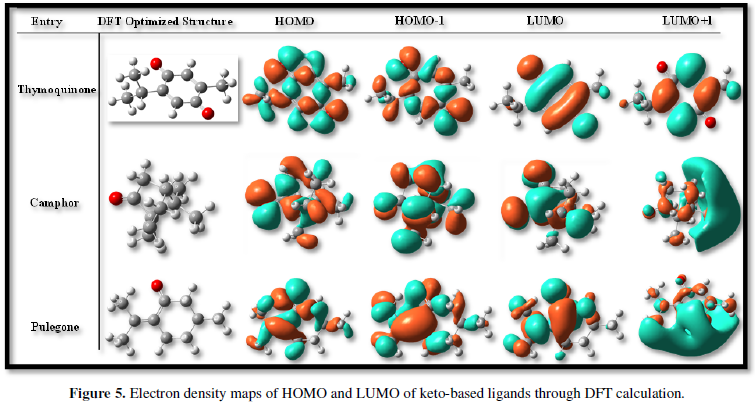
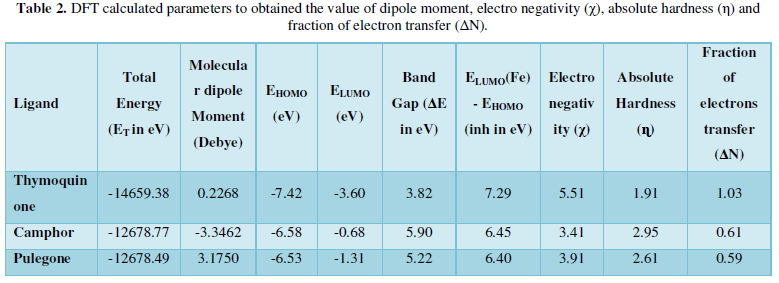
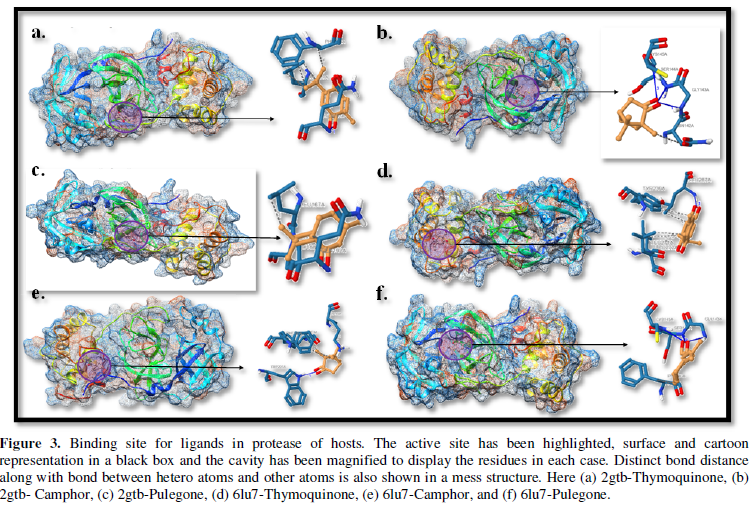
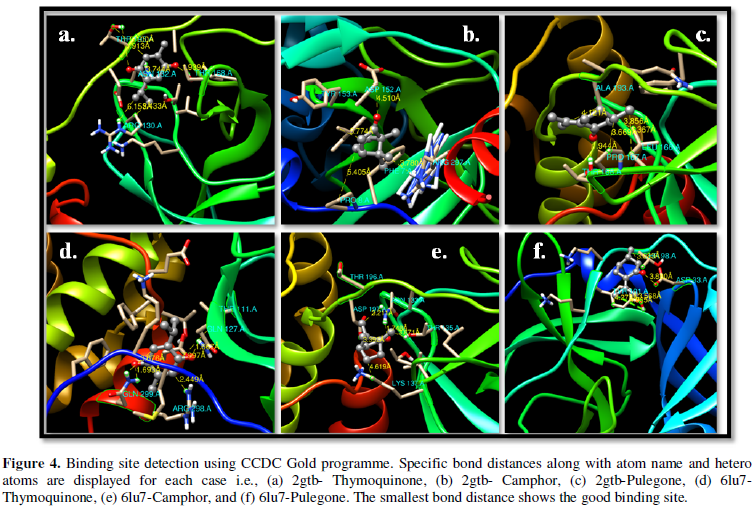
COVID-19, produced by SARS-CoV-2 has just inaugurated as a global pandemic. Intense efforts are also underway to discover a drug or vaccine across the globe to resist the outbreak. Nowadays, alternative treatments for the management of the disease are also being explored. It has been proven that phytochemicals from various plant extract are the promising candidate in dealing many therapeutic activities with minimum side effects. Herein, this article will cover the major uses of keto based natural drugs such as pulegone, camphor, thymoquinone for their antimicrobial properties against the proteases of receptor binding domain, which is one of the vital targets for novel antiviral agents. In order to discover the new possible COVID-19 inhibitors, the proteases PDBs e.g., 2gtb and 6lu7 will be used as hosts to calculate the interactions with those aforementioned natural drugs as guests. Those protein compounds are chosen because they share 96% similarity of antiviral nature. A comprehensive molecular docking approach (through both Auto dock and CCDC GOLD) have been introduced for the theoretical calculations of binding energies as a result of host-guest interactions. Furthermore, protein ligand interaction profiler (PLIP) server also provides hydrophobic interactions (clear data about amino acid residues along with bond distances), 3D view of molecules, number of hydrogen bonds, etc. Present research shows that pulegone is the most active having binding energy -5.93 kcal/ mol against 2gtb viral spike-protein, whereas, on the other, that of camphor is -4.92 kcal/ mol against 6lu7 protease of SARS-CoV-2.
- Wu F, Zhao S, Yu B, Chen YM, Wang W, et al. (2020) A new coronavirus associated with human respiratory disease in China. Nature 579: 265-269.
- Zhu N, Zhang D, Wang W, Li X, Yang B, et al. (2020) A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382(8): 727-733.
- Hui DS, Azhar EI, Madani TA, Ntoumi F, Kock R, et al. (2020) The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health - The latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis 91: 264-266.
- WHO (2020) Mental health and psychosocial considerations during the COVID-19 outbreak. Available online at: https://www.who.int/docs/default-source/coronaviruse/mental-health-considerations.pdf
- Swamy MK, Akhtar MS, Sinniah UR (2016) Antimicrobial properties of plant essential oils against human pathogens and their mode of action: An updated review. Evid Based Complement Alternat Med 2016: 3012462.
- Valdivieso-Ugarte M, Gomez-Llorente C, Plaza-Díaz J, Gil A (2019) Antimicrobial, antioxidant, and immunomodulatory properties of essential oils: A systematic review. Nutrients 11(11): 2786.
- Pandey AK, Kumar P, Singh P, Tripathi NN, Bajpai VK (2017) Essential oils: Sources of antimicrobials and food preservatives. Front Microbiol 7: 2161.
- Pourghanbari G, Nili H, Moattari A, Mohammadi A, Iraji A (2016) Antiviral activity of the oseltamivir and Melissa officinalis L. essential oil against avian influenza A virus (H9N2). Virus Dis 27: 170-178.
- Tariq S, Wani S, Rasool W, Shafi K, Bhat MA, et al. (2019) A comprehensive review of the antibacterial, antifungal and antiviral potential of essential oils and their chemical constituents against drug-resistant microbial pathogens. Microb Pathog 134: 103580.
- Srivastava P, Tiwari A (2017) Critical role of computer simulations in drug discovery and development. Curr Top Med Chem 17: 2422-2432.
- Ekins S, Mestres J, Testa B (2007) In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br J Pharmacol 152: 9-20.
- Terstappen GC, Reggiani A (2001) In silico research in drug discovery. Trends Pharmacol Sci 22: 23-26.
- Kundu BK, Pragti, Mobin SM, Mukhopadhyay S (2020) Studies on the influence of the nuclearity of zinc(ii) hemi-salen complexes on some pivotal biological applications. Dalton Trans 49: 15481-15503.
- Majumdar D, Philip JE, Das S, Kundu BK, Saini RV, et al. (2021) Experimental and theoretical corroboration of antimicrobial and anticancer activities of two pseudohalides induced structurally diverse Cd (II)-Salen complexes. J Mol Struct 1225: 129189.
- Das M, Biswas A, Kundu BK, Charmier MAJ, Mukherjee A, et al. (2019) Enhanced pseudo-halide promoted corrosion inhibition by biologically active zinc (II) Schiff base complexes. Chem Eng J 357: 447-457.
- Das M, Biswas A, Kundu BK, Mobin S, Udayabhanu G, et al. (2017) Targeted synthesis of cadmium(ii) Schiff base complexes towards corrosion inhibition on mild steel. RSC Adv 7: 48569-48585.
- Das M, Kundu BK, Tiwari R, Mandal P, Nayak D, et al. (2018) Investigation on chemical protease, nuclease and catecholase activity of two copper complexes with flexidentate Schiff base ligands. Inorg Chim Acta 469: 111-122.
- Kundu BK (2019) Studies on metal complexes of 'N, O-Donor' ligands in modelling metalloenzymes sensing and catalysis, PhD Thesis, IIT Indore.
- Mandal P, Kundu BK, Vyas K, Sabu V, Helen A, (2018) Ruthenium(ii) arene NSAID complexes: inhibition of cyclooxygenase and antiproliferative activity against cancer cell lines. Dalton Trans 47: 517-527.
- Mandal P, Malviya N, Kundu BK, Singh DS, Nagaraja CM, et al. (2017) RAPTA complexes containing N‐substituted Tetrazole scaffolds: Synthesis, characterization and Antiproliferative activity. Appl Organomet Chem 32: 4179-4191.
- Kundu BK, Chhabra V, Malviya N, Ganguly R, Mishra GS, et al. (2018) Zeolite encapsulated host-guest Cu(II) Schiff base complexes: Superior activity towards oxidation reactions over homogenous catalytic systems. Micropor Mesopor Mat 271: 100-117.
- Kundu BK, Das M, Ganguly R, Bhobe PA, Mukhopadhyay S (2020) Role of zeolite encapsulated Cu(II) complexes in electron transfer as well as peroxy radical intermediates formation during oxidation of thioanizole. J Catal 389: 305-316.
- Kundu BK, Pragti, Biswas S, Mondal A, Mazumdar S, et al. (2021) Unveiling the urease like intrinsic catalytic activities of two dinuclear nickel complexes towards the in-situ syntheses of amino cyanopyridines, Dalton Trans.
- Kundu BK, Ranjan R, Mukherjee A, Mobin SM, Mukhopadhyay S (2019) Mannich base Cu(II) complexes as biomimetic oxidative catalyst. J Inorg Biochem 195: 164-173.
- Chhabra V, Kundu BK, Ranjan R, Pragti, Mobin SM, et al. (2020) Coligand driven efficiency of catecholase activity and proteins binding study of redox active copper complexes. Inorg Chim Acta 502: 119389.
- Kundu BK, Mandal P, Mukhopadhyay BG, Tiwari R, Nayak D, et al. (2019) Substituent dependent sensing behavior of Schiff base chemosensors in detecting Zn2+and Al3+ ions: Drug sample analysis and living cell imaging. Sens Actuators B: Chem 282: 347-358.
- Kundu BK, Pragti, Reena, Mobin SM, Mukhopadhyay S (2019) Mechanistic and thermodynamic aspects of a pyrene-based fluorescent probe to detect picric acid. New J Chem 43: 11483-11492.
- Kundu BK, Singh R, Tiwari R, Nayak D, Mukhopadhyay S (2019) An amide probe as a selective Al3+ and Fe3+ sensor inside the HeLa and a549 cell lines: Pictet-Spengler reaction for the rapid detection of tryptophan amino acid. New J Chem 43: 4867-4877.
- Zhang L, Lin D, Sun X, Curth U, Drosten C, et al. (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368: 409-412.
- Lan J, Ge J, Yu J, Shan S, Zhou H, et al. (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581: 215-220.
- Xu Z, Peng C, Shi Y, Zhu Z, Mu K, et al. (2020) Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation, BioRxiv.
- Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, et al. (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181: 281-292.
- Trott O, Olson A (2010) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31: 455-461.
- Dallakyan S, Olson AJ (2015) Small-molecule library screening by docking with PyRx, in: Chemical biology, Springer. pp: 243-250.
- O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, et al. (2011) Open Babel: An open chemical toolbox. J Chem Inform 3: 1-14.
- Geerlings P, Chamorro E, Chattaraj PK, De Proft F, Gázquez JL, et al. (2020) Conceptual density functional theory: Status, prospects, issues. Theor Chem Acc 139: 1-18.
- Leach AR, Shoichet BK, Peishoff C (2006) Prediction of protein - ligand interactions. Docking and scoring: Successes and gaps. J Med Chem 49: 5851-5855.
- Vieira TF, Sousa SF (2019) Comparing AutoDock and Vina in ligand/decoy discrimination for virtual screening. Appl Sci 9: 4538.
- Elfiky AA, Ismail AM, Elshemey WM (2020) Recognition of gluconeogenic enzymes; Icl1, Fbp1, and Mdh2 by Gid4 ligase: A molecular docking study. J Mol Recognit 33: e2831.
- Leach AR (2001) Molecular modelling: Principles and applications. Pearson Education.
- Elfiky AA (2020) Novel guanosine derivatives against Zika virus polymerase in silico. J Med Virol 92: 11-16.
- Bostan R, Varvara S, Găină L, Mureşan LM (2012) Evaluation of some phenothiazine derivatives as corrosion inhibitors for bronze in weakly acidic solution. Corros Sci 63: 275-286.
-
 Table 1
Table 1 -
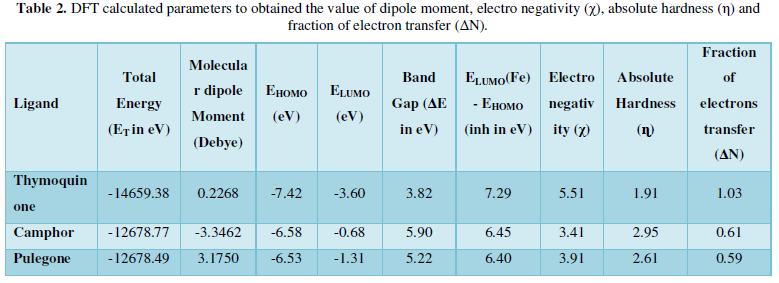
Table 2




QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Renal Transplantation Science (ISSN:2640-0847)
- Stem Cell Research and Therapeutics (ISSN:2474-4646)
- Dermatology Clinics and Research (ISSN:2380-5609)
- Journal of Alcoholism Clinical Research
- Journal of Cell Signaling & Damage-Associated Molecular Patterns
- International Journal of Clinical Case Studies and Reports (ISSN:2641-5771)
- Journal of Spine Diseases


