3106
Views & Citations2106
Likes & Shares
Abbreviations: MDSCs: Skeletal
Muscle-Derived Stem Cells, Sk-SCs:
Skeletal Muscle-Derived Multipotent Stem Cells, Sk-34: CD45-/34+, Sk-DN: CD45-/34-, GFP: Green Fluorescent Protein, DMEM: Dulbecco’s Modified Eagle’s Medium, FCS: Fetal Calf Serum, bFGF:
Basic Fibroblast Growth Factor, PCR:
Polymerase Chain Reaction PBS: Phosphate Buffered Saline, PFA: Paraformaldehyde, PB: Phosphate
Buffer, EDTA: Ethylene Diamine Tetra Acetic Acid, ALP: Alkaline Phosphatase,
BMSCs: Bone-Marrow Stromal Cells, DAPI: 4,6-Diamino-2-Phenylindole, BMP: Bone
Morphogenetic Protein, FBS: Fetal Bovine Serum.
The osteogenic differentiation potential of mouse skeletal
muscle-derived stem cells, sorted as CD34+/45- (Sk-34)
and CD34-/45- (Sk-DN), was examined by direct
transplantation in an experimental tibial bone fracture model. Freshly isolated
Sk-34 and Sk-DN cells cultured for 5 days were obtained from GFP
transgenic-mice using mild enzymatic treatment, without any induction of osteogenic
differentiation. Cell pellets were directly implanted into the fractured tibial
bone model generated by drilling-removal of 1/2 the circumference of the bone
matrix. Concurrently, transplantations of muscle-derived CD45+ cells
(residual population after sorting of Sk-34 and Sk-DN cells), bone-marrow
stromal cells (BMSCs), and medium without any cells, were also performed as
control experiments. After 2 months, favorable bone healing was achieved in all
5 groups, suggesting the possibility of natural healing in the present model.
However, active engraftment of GFP+ cells was observed in Sk-34
(9/9) and Sk-DN (6/9) group by macroscopic fluorescence stereomicroscopy, but
no GFP+ cells were detected in the other 2 groups.
Immunohistochemical analysis showed frequent presence of GFP+/osteocalcin+
cells (putative osteoblasts) in the bone matrix of the Sk-34 group. The same
trend was also observed in the Sk-DN and BMSC group, but the detectable number
of these cells was relatively lower than that observed with Sk-34. At 6 months
after transplantation, GFP+ donor cells with a line-cell like
structure, located on the inner surface of the bone matrix were still observed
in the Sk-34 group. A similar trend was also observed in both the Sk-DN and
BMSC groups, but GFP+ cells were fewer in number in these groups.
These results indicate that Sk-34 and Sk-DN cells can differentiate into
osteoblasts in vivo following bone fracture, in a manner similar to BMSCs. They
exert their original “milieu-dependent differentiation capacity” without being
inducted to the osteogenic lineage ex vivo, and are committed to the bone
re-modeling cycle even after 6 months of transplantation.
INTRODUCTION
The bone-autograft is the most common method to heal large-size bone defects in orthopedic therapy, whereas an additional invasive surgical procedure may lead to donor site morbidity. Therefore, it is difficult to obtain a sufficient volume of bone autograft for a critically large-sized defect. In this case, although allografts have been considered, there are concerns about disease transmission and immune rejection. To overcome this problem, a large number of synthetic grafts such as metallic implants, ceramics, polymers, and composites of these materials have been developed [1,2]. However, the problem with these is the absence of osteogenicity (supply of bone-forming cells), osteoinductivity (initiation of cell differentiation), and osteocondutivity (facilitation of cell and nutrient infiltration).
Consequently, the recent trend is focused on calcium
phosphate-containing synthetic biomaterials such as hydroxyapatite relating
substances which show properties of osteoinductivity [3], but the results of
their sole use are still not satisfactory. For this purpose, fabricated
synthetic biomaterials or scaffolds co-transplanted with osteogenic stem cells,
were considered to be capable grafts with all three biological capacities
mentioned above.
Several stem cells including bone marrow stromal cells [4], adipose
tissue-derived stem cells [1], and skeletal muscle-derived stem cells (MDSCs)
have been used for healing bone defects. However, from the clinical point of
view, heterotopic ossification in the skeletal muscle has been well-known as an
example of ectopic bone formation in the soft-tissue [5,6] . In this context,
stem, and/or progenitor cells from the skeletal muscle are likely to have
somewhat of an advantage than the others. In fact, accelerated bone formation
after MDSC transplantations has been reported [7,8], whereas the level of their
commitment to bone formation, such as a detailed analysis of the fate and
differentiation of the engrafted cells is still not clear.
We also fractionated mouse skeletal muscle-derived multipotent stem
cells (Sk-SCs), as CD45-/34+ (Sk-34) and CD45-/34-
(Sk-DN or Sk-DN/29+) cells, using original and mild enzymatic
isolation and fluorescence activated cell sorting (FACS) [9,10]. These cells
were capable of synchronized reconstitution of the muscle-nerve-blood vessel
unit associated with the capacity to differentiate into skeletal muscle cells,
Schwann cells, perineurial/endoneurial cells, pericytes, vascular smooth muscle,
and endothelial cells after in vivo transplantation into various tissues
[11-16]. We also fractionated the human skeletal muscle-derived Sk-34 and
Sk-DN/29+ cells using the same method as above, and demonstrated
that they have comparable differentiation and tissue reconstitution capacities
to mouse cells [17]. However, the osteogenic capacity of Sk-34 and Sk-DN cells
is still unknown as was observed in MDSCs derived from the pre-plating culture
system derived [7,8].
Therefore, the purpose of this study is to clarify whether mouse Sk-34
and Sk-DN cells can differentiate into osteogenic cells, and facilitate bone
formation after in vivo transplantation. For this purpose, we developed the
bone defect model in the tibia, and demonstrated that both Sk-34 and Sk-DN
cells upon transplanting, differentiated into osteoblastic cells and remained
committed to the bone metabolic cycle for over 6 months.
Materials and
Methods
Animals
Green fluorescent protein transgenic mice (GFP-Tg mice; C57BL/6 TgN[act
EGFP]Osb Y01, provided by Dr. M. Okabe, Osaka University, Osaka, Japan) [18]
were used as donor mice (male, 4-8 week-old, n=12), and wild-type mice
(C57BL/6N) were used as recipients (male, 8-12 week-old, n=29) in the
transplantation experiments. All experimental procedures were approved by the
Tokai University School of Medicine Committee on Animal Care and Use.
Purification and preparation of transplanted
Cells
Muscle sampling was performed under an overdose of pentobarbital (60
mg/kg, Schering-Plough, + butorphanol tartrate 2 mg/kg, Meiji Seika, Tokyo,
Japan, i.p.). The thigh and lower leg muscles (tibialis anterior, extensor
digitorum longus, soleus, plantaris, gastrocnemius, and quadriceps femoris) of
GFP-Tg mice were removed and used for subsequent experiments. The average total
muscle mass removed was 509 ± 47 mg/GFP-Tg mouse (mean ± SE). Sk-SCs were
isolated according to the previously described procedure [9,10]. Briefly,
muscles were not minced, and were then treated with 0.1% collagenase type IA
(Sigma-Aldrich, Tokyo, Japan) in Dulbecco’s modified Eagle’s medium (DMEM,
Wako, Osaka, Japan) containing 7.5% fetal calf serum (FCS, Equitech Bio, TX,
USA) with gentle agitation for 2 h at 37°C. After digestion, isolated cells
were serially filtered through 70-, 40-, and 20-μm nylon filters, in that
order, to remove debris. Cells were stained against CD45 and CD34, and sorted
as Sk-34 (CD45-/34+), Sk-DN (CD45-/34-)
cells and CD45+ cells. Sk-34 and CD45+ cells were freshly
prepared for the transplantation experiment. However, Sk-DN cells were expanded
in a collagen-based culture medium (CollagenCult H4742, StemCell Tech.,
Vancouver, Canada) with 10 ng/ml bFGF and 20 ng/ml EGF for 5 days before the
transplantation, because of their small number and immaturity [10,19,20].
In addition, whole bone-marrow stromal cells (BMSCs) were obtained by
flushing the tibias and femurs of GFP mice (n=2-3/experiment). After
elimination of red blood cells, mononuclear bone marrow cells were obtained,
and cultured in 20%FCS/DMEM for 48 hours. Floating cells were eliminated and
the remaining adhesive cells were cultured further for 5 days (total of 7
days). Media were changed after every 2 days for both cultures. Expanded BMSCs
were transplanted in the same manner as both Sk-SCs.
Furthermore, CD45+ cells, which were sorted at the same time
for other two Sk-SCs and were considered as contaminated hematopoietic cells
from circulating blood, were also transplanted as a control group.
Transplantation of the same amount of media without cells was also performed to
evaluate the present bone fracture model. Consequently, we transplanted 5 types
of cells: (1) freshly isolated Sk-34 cells (n=9), (2) freshly isolated CD45+
cells (n=4), (3) expanded Sk-DN cells (n=9), (4) expanded BMSCs (n=4), and (5)
medium without cells (n=3).
RT-PCR for Sk-MSCs before and after transplantation
In order to test the expression of specific
markers of cell differentiation and paracrine capacities as the putative
indicators of osteogenic cells, bulk cell RT-PCR was performed. Specific
primers and the materials analyzed are summarized in Table 1. Just prior to transplantation, some cells were lysed and
total RNA was purified using a QIAGEN RNeasy micro kit. First-strand cDNA
synthesis was performed with an Invitrogen SuperScript III system using
dT30-containing primers (see above), and specific PCR (35 cycles of 30 seconds
at 94°C, 30 seconds at 60-65°C and 2 minutes at 72°C) was performed in a 15-µl reaction containing
ExTaq buffer, 0.8 U of ExTaq-HS-polymerase, 0.7 µM specific sense and antisense
primers,
Preparation of the bone defect model on the
tibia
In order to generate the experimental bone defect, the left tibia of
the mice was exposed after skin incision and blunt dissection of the periosteum
and muscles. This was followed by an intramedullary nailing from the proximal
end using a 27G syringe needle. After retraction of the skin and the muscles, a
square-hole (2 x 2mm) was bored with an electric drill occupying almost 1/2 of
the whole circumference (Figure 1B).
The hole was then filled with the medium containing each above cell type (3-4µl
with 1x 105 cells/µl, Figure
1A). The transplanted portion was covered by replacing the periosteum and
muscles, and suturing the skin.
Macroscopic Observation and Immunostaining
At 2 and 6 months after transplantation, recipient mice were given an overdose of pentobarbital (60 mg/kg, i.p) + xylazine HCl (10 mg/kg, i.p.), and the engraftment of donor-derived GFP+ cells in the damaged portion of the tibial bone was confirmed by fluorescence stereomicroscopy (SZX12; Olympus, Tokyo, Japan) (see Figure 3A). Recipient mice were then perfused with warm 0.01 M phosphate buffered saline (PBS) through the left ventricle, followed by fixation with 4% paraformaldehyde/0.1 M phosphate buffer (4% PFA/PB). The tibial bone was then removed and fixed overnight in 4% PFA/PB at 4°C followed by washing with a graded sucrose (0–25%)/0.01 M PBS series continuously containing 0.25M EDTA during the course of 1 week. Samples were then immersed in OCT compound and frozen/stored at -80°C. Subsequently, 7-µm histological sections were prepared. Localization of osteoblasts was detected by goat polyclonal anti-osteocalcin (dilution = 1:1800; incubation=4°C overnight; Biomedical technologies, Stoughton, MA), and alkaline phosphatase staining (ALP staining kit, Mutoh chemical, Tokyo, Japan). Reactions were visualized using Alexa Fluor-594-conjugated rabbit anti-goat antibodies (1:500, room temperature for 2 h; Molecular Probes, Eugene, OR). Nuclei were counter-stained with DAPI (4,6-diamino-2-phenylindole).
RESULTS
Evaluation of the experimental bone fracture
model
Favorable healing of bone fracture was observed in the medium control group, and so this model can be considered as a naturally healing model. In this context, this experiment is a confirmation study of how transplanted cells may contribute and/or commit to bone formation following a natural healing process.
Putative osteogenic capacity of Sk-34, Sk-DN
cells, and BMSCs just before transplantation
The putative osteogenic capacity of Sk-34, Sk-DN, and BMSCs just before
transplantation was determined by RT-PCR analysis (Figure 2). The mRNA expression of seven markers specific for
osteogenic cells was examined in freshly isolated Sk-34 cells, Sk-DN cells
cultured for 5 days, and BMSCs expanded for 7 days. Results indicate that all
three cells showed almost equal expression of all markers except for alkaline
phosphatase (lane No. 4), which was lacking in Sk-DN cells. Therefore, all
three cells may show putative osteogenic capacities before transplantation that
seems relatively higher in Sk-34 cells and BMSCs.
Macroscopic fluorescence stereomicroscopy
Macroscopic fluorescence stereomicroscopy showed no GFP+
emissions in the BMSCs and CD45+ transplanted bones, even though
bone healing was complete. However, in the Sk-34 and Sk-DN cell-transplanted
groups, clear GFP emissions were consistently observed. Typical engraftment of
GFP+ tissues in the tibial bone at 2 months after Sk-34 and Sk-DN
cell transplantations is shown in Figure
3A and 3B. The damaged portion showed a thicker circumference than the
normal portion, and GFP+ cells were strictly incorporated in the
bone tissues of the recipient (arrows). Detection of GFP+
tissue-engraftment was observed in 9/9 instances of Sk-34 and in 6/9 instances
of Sk-DN transplantation. In addition, the Sk-DN transplantation group a showed
relatively smaller volume of GFP+ tissues compared to the Sk-34
group.
Histological analysis
Histological analysis of the damaged tibial portion in Sk-34 cell transplantation is shown in Figure 3C and D. Immunohistochemical detection using anti-osteocalcin clearly indicated that several GFP+ cells in the bone matrix close to the marrow are osteocalcin+ (arrows in Fig. 3C). These are considered as early stage osteocytes, suggesting that the transplanted donor Sk-34 cells could differentiate into osteoblasts and contribute to bone matrix formation. Similarly, several GFP+ cells also showed alkaline phosphatase positive reactions in the marrow area, thus providing further evidence of osteoblast differentiation (arrows in Figure 3D).
Similar patterns were also observed in the Sk-DN cell and BMSCs
transplantation group, but the number of cells involved in these was relatively
fewer than Sk-34, as revealed by the macroscopic observations above.
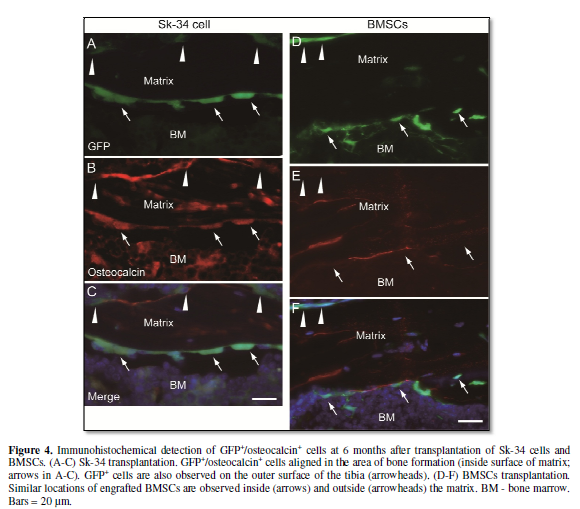
Furthermore, GFP+/osteocalcin+ cells were detected even at 6 months after transplantation. Donor-derived osteocalcin+ and GFP+ cells lined up on the border between the bone matrix and marrow (Figure 4A-C), thus considered as pre-osteoblasts. A similar trend but with few cells was observed in the Sk-DN group. This result suggests that donor-derived GFP+ cells can continuously reside for 6 months in vivo, and participate in the bone metabolic cycle or in the bone re-modeling process. In fact, comparable behavior such as formation of GFP+ bone lining-cell like structures (negative for osteocalcin), was also observed in BMSC transplantation at 6 months (Figure 4D-F), although the corresponding detectable area was smaller than in Sk-34 cell transplantation.
DISCUSSION
The present study clearly demonstrated that
Sk-SCs, which were sorted as Sk-34 and Sk-DN cells, differentiated into
osteoblasts after direct and separate transplantation into fractured bone. In
addition, the present result also demonstrated that engrafted Sk-34 cells
aligned serially on the inner surface of the bone matrix (bone formation area)
were evident even after 6 months of transplantation (Figure 4A-C). This indicates that the engrafted donor cells thrive
in the bone formation area of the cavity in a manner similar to typical
pre-osteoblasts derived from the marrow stroma [21], and a similar trend was
also confirmed by BMSCs transplantation in the present study (Figure 4D-F). In case the transplanted
cells were to show only a single episode of bone formation during the
early-stage of regeneration, they would be eliminated over 6 months. However,
the engrafted cells were retained in the bone and showed a preserved
pre-osteogenic aligning structure. This suggested the commitment of
transplanted cells into the ossification cycle during 6 months.
The applications of skeletal muscle-derived stem cells in the bone
healing process have been previously reported [7,22]. However, these studies
used ex vivo activation for the cells, with agents such as bone morphogenetic
protein (BMP) -2, -4 -6 and -7 [23-27]. Similar methods were also applied to
other stem cells, such as fibroblasts [28], bone marrow derived cells [29,30],
and fat derived cells [31,32]. However, the most appropriate cell type for bone
healing is still unknown. In this study, we did not subject the Sk-34 and Sk-DN
cells to any ex vivo induction toward the osteogenic lineage, but the
expression of mRNAs specific to the bone lineage was naturally observed in
these cells before transplantation (Figure
2). In addition, osteogenic differentiation was not detected in the
expansion cell culture system (data not shown). This indicates that the actual
osteogenic differentiation was mainly induced after in vivo transplantation due
to “milieu dependent differentiation”. The microenvironment of fractured bone
naturally comprises various ossification related factors (cytokines,
chemokines, growth factors and hormones), which may naturally induce osteogenic
differentiation of the transplanted Sk-SCs. To our knowledge, this is the first
report of such “milieu dependent osteogenic differentiation” and “the
commitment to the ossification cycle” in Sk-SCs. Therefore, we hypothesize that
osteogenic differentiation potential is higher in Sk-34 and Sk-DN cells than
other cells used in the present study.
With respect to the differences between the Sk-SCs used in the present
study and other skeletal muscle-derived stem cells, particular consideration
was given to the cell isolation method. During enzymatic digestion, proteolytic
contamination can damage cell-surface ligands or receptors, which are necessary
for stem cell function after in vivo transplantation [33]. Therefore, we have
consistently used lower concentrations of collagenase (0.1%) in DMEM followed
by addition of 5-10% FBS to the collagenase solution in order to minimize
contaminating protease activity and to protect the isolated cells. This is a
mild treatment when compared with those used in previous studies, which used
higher collagenase concentrations (0.2-2.0%) without suppression of protease
activity, followed by dispase (0.25%) and/or trypsin (0.1-0.25%) treatments.
Furthermore, in contrast to other reports, intact donor muscle was used for
enzymatic digestion instead of minced tissue. These factors mostly affected in
vivo differentiation potential. In addition, we recently isolated human Sk-SCs
using the same method and found that they had a similar differentiation
capacity as their mouse counterparts [17]. However, human Sk-SCs can be divided
two particular cell populations; the Sk-DN fraction showed limited inclusion of
skeletal-myogenic cells, whereas, the remaining multipotent stem cells were
contained in the Sk-34 fraction [17]. Therefore, which cells can be
differentiate into osteogenic cells is still unknown but interesting.
Concerning the lower engraftment ratio in Sk-DN cells, mouse Sk-DN
cells are placed upstream to Sk-34 cells in stem cell hierarchy [20], and exert
the same differentiation and tissue reconstitution capacities in damaged
muscle-nerve-blood vessel units [15,19]. However, due to the lesser number of
freshly isolated Sk-DN cells, culture expansion was necessary in this
experiment. This expansion process may induce relatively higher
skeletal-myogenesis in Sk-DN cells than in Sk-34, and may result in lowering
the osteogenic potential of Sk-DN cells. This suggests that the process of
skeletal-myogenesis is reciprocal to that of osteogenesis. In other words,
inhibition of skeletal-myogenesis may induce osteogenesis in Sk-SCs.
However, critically large-sized bone defects, the combined use of
scaffolds may be absolutely necessary. Therefore, materials showing affinity
for cell adherence are considered to be the best sources of scaffolds. In this
context, hydroxyapatite and β-tricalcium phosphate may be good candidate for
treating bone fractures [1,3,34,35]. We are currently investigating the
combined use of hydroxyapatite and Sk-SCs for critically large bone fractures
and their capacity for bone formation.
CONCLUSION
The present data indicates that mouse Sk-SCs,
sorted as Sk-34 and Sk-DN cells, differentiated into osteoblasts in the in vivo
bone fracture niche after 2 months of transplantation. These cells were
retained even after 6 months of transplantation and participated in the bone
re-modeling cycle, as efficiently as transplanted BMSCs.
-
 Table 1
Table 1




QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Forensic Research and Criminal Investigation (ISSN: 2640-0846)
- Journal of Alcoholism Clinical Research
- International Journal of Surgery and Invasive Procedures (ISSN:2640-0820)
- Ophthalmology Clinics and Research (ISSN:2638-115X)
- International Journal of Anaesthesia and Research (ISSN:2641-399X)
- Oncology Clinics and Research (ISSN: 2643-055X)
- Journal of Spine Diseases


