947
Views & Citations10
Likes & Shares
INTRODUCTION
Polyhydramnios is identified in 1-2% of pregnancies [1]. It can result in the second trimester as the fetal kidneys become the predominant source of amniotic fluid. Polyhydramnios may be related to maternal or fetal conditions including gestational diabetes, congenital infection, congenital anomalies and genetic disorders [2]. Evaluation typically includes maternal testing for diabetes, targeted anatomic ultrasound, maternal serology for infectious diseases and occasionally offering genetic amniocentesis. Despite thorough investigation, 50-60% of cases will be idiopathic [3]. In 10% of these cases, an anomaly will only be found after birth [4]. Polyhydramnios has been associated with adverse pregnancy outcomes, including fetal death, preterm delivery and increased neonatal morbidity [5]. There are only a few genetic diseases associated with polyhydramnios, including antenatal Bartter syndrome.
Bartter syndrome is a rare autosomal recessive salt losing tubulopathy first described in 1962 [6]. It is caused by defective salt reabsorption in the thick ascending loop of Henle [7]. It can present in two ways: antenatal and classical Bartter syndrome. The classic form has insidious onset in infancy; children present with severe polyuria, dehydration, hypokalemic metabolic alkalosis and failure to thrive [8]. Typically, the prenatal presentation is very severe with marked polyhydramnios, fetal growth restriction, preterm delivery, postnatal polyuria, dehydration, hypokalemic metabolic alkalosis, marked hypercalciuria leading to nephrocalcinosis and failure to thrive [9,10]. Because symptoms at birth can be life threatening, prenatal diagnosis is important for management both prenatally and in the immediate postnatal period.
An accurate diagnosis is an integral component of patient care for children with rare genetic disorders. However, precise diagnosis of an underlying genetic defect is often challenging as most cases lack a family history and predisposing factors. Currently, prenatal testing for a sonographically identified abnormality includes fluorescence in situ hybridization for rapid aneuploidy testing, G-banded karyotyping and chromosomal microarray analysis for detecting copy number variants. These cytogenetic tests are limited by their resolution. If these techniques are used in conjunction in fetuses with structural abnormalities, they will identify an underlying etiology in up to 40% of dysmorphic fetuses [11]. This leaves a majority of cases without a diagnosis, particularly in the absence of a structural abnormality. Significant advances in next generation sequencing technology over the recent years, particularly whole exome sequencing (WES), have revolutionized the practice in genetic diagnosis. WES sequences ~95% of the genome; specifically, it examines the protein-coding exons of ~20,000 genes. Although exons encompass less than 2% of the genome, they contain >85% of all disease causing pathogenic variants [11]. WES has recently become an accepted test in the neonatal, pediatric and adult populations. Postnatal WES in patients with a suspected genetic abnormality who remain undiagnosed with traditional methods has a diagnostic yield of 25-30% [12]. In recent years, WES is being used for identify pathogenic variants in fetuses with structural abnormalities [13]. The ability to diagnose pathogenic single nucleotide variants prenatally has been limited to a small number of genetic diseases with recognizable phenotypes [14]. Various single gene disorders present prenatally and are undiagnosed with traditional cytogenetic techniques, many of which may be diagnosed using WES.
All patients that are considered for WES should receive pre- and post-test counseling from a provider with genetics expertise [15]. As WES is a newer modality for prenatal testing and studies are ongoing for its clinical utility, The American College of Medical Genetics and Genomics (ACMG) and Society of Maternal-fetal Medicine (SMFM) do not recommend routine use for prenatal diagnosis [16]. In those cases in which other approaches to diagnosis are uninformative, it may be appropriate to offer WES. Sequencing of the whole exome and protein-coding regions is well justified as an efficient approach to diagnose variants underlying rare genetic conditions. It can improve the accuracy of prognosis, prenatal counseling and pregnancy management, while reducing the diagnostic testing that occurs postnatally. This may result in a reduction of the costs of molecular, imaging and other investigations. It can also improve early postnatal management in cases of life threatening neonatal conditions, such as Bartter syndrome [13,17,18].
CASE REPORT
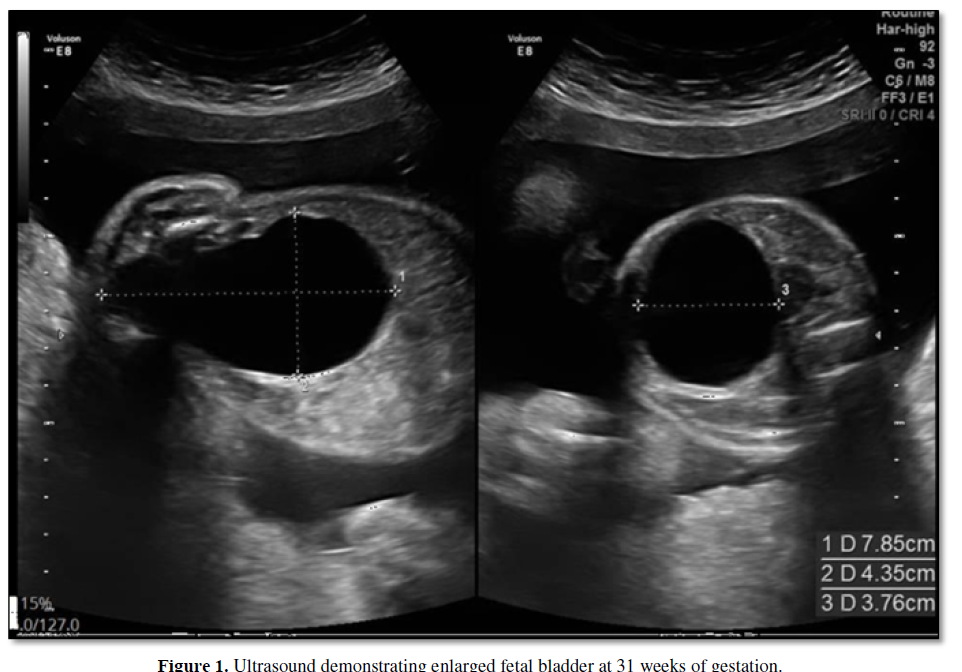
We report on a 33 year old Arabic woman in her third pregnancy (G3P2002). She had a history of gestational diabetes, which was being treated with Metformin. She did not report any significant family history. Fetal anatomy survey at 23 weeks revealed an enlarged fetal bladder (Figure 1); amniotic fluid was assessed as normal. Due to the enlarged fetal bladder, a follow up ultrasound was performed at 27 weeks gestation; this revealed a normally grown fetus and polyhydramnios (amniotic fluid volume (AFI) 44 cm, with the deepest fluid pocket of 14 cm) in the absence of any other structural abnormality. Patient presented at 31 weeks with abdominal discomfort and fetus was then noted to be growth restricted (EFW 1369 g, <5th percentile) with normal Doppler studies and AFI 65 cm. After discussion of therapeutic options, serial amnioreductions were performed, removing approximately 3 L of amniotic fluid each time.
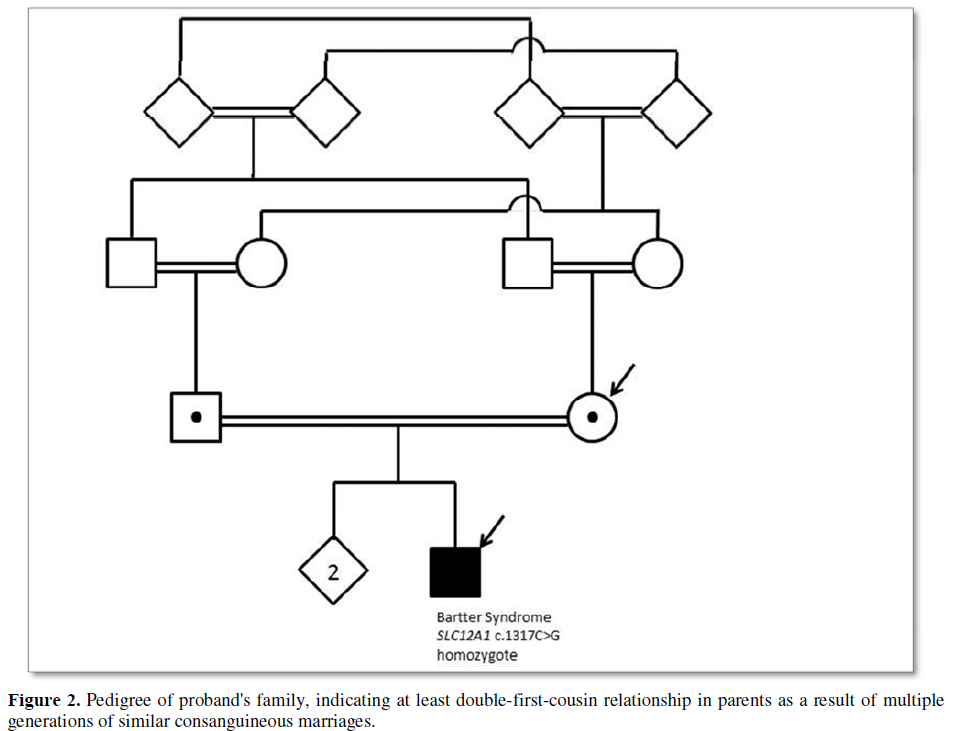
Fetal karyotype performed on cultured amniocytes revealed normal male chromosomes (46, XY). Chromosomal microarray analysis (180K SNP-aCGH array) revealed no clinically significant copy number variants; however, the SNP array discovered several regions of homozygosity encompassing 12.1% of the genome, indicating inheritance from a shared ancestor. A family history was further reviewed with the patient after these findings and she reported being double-first cousins with her husband (Figure 2). A fetal MRI was also performed to assess the upper airway of the fetus, which appeared normal. The pregnancy continued with weekly Doppler studies and bi-weekly non-stress tests.
At 32 weeks of gestation, the umbilical artery demonstrated intermittent absent end diastolic flow and the patient received two doses of betamethasone. A repeat Cesarean section delivery was performed secondary to fetal bradycardia at 33 weeks of gestation. The patient delivered a male neonate with a birth weight of 1590 g and APGAR scores of 8 at 1 min and 9 at 5 min of age. The neonate was noted to have polyuria and severe electrolyte disturbances soon after birth (urine output 8 mL/kg/h, sodium 151, potassium 6.3, anion gap 26). These abnormalities were thought to be secondary to immature fetal kidneys, which were expected to normalize over the next several days. The neonate was started on feeds and maintenance fluid, supplementing with sodium and potassium; the urine output normalized by 2 weeks of age. The neonate was discharged home at 6 weeks of life.
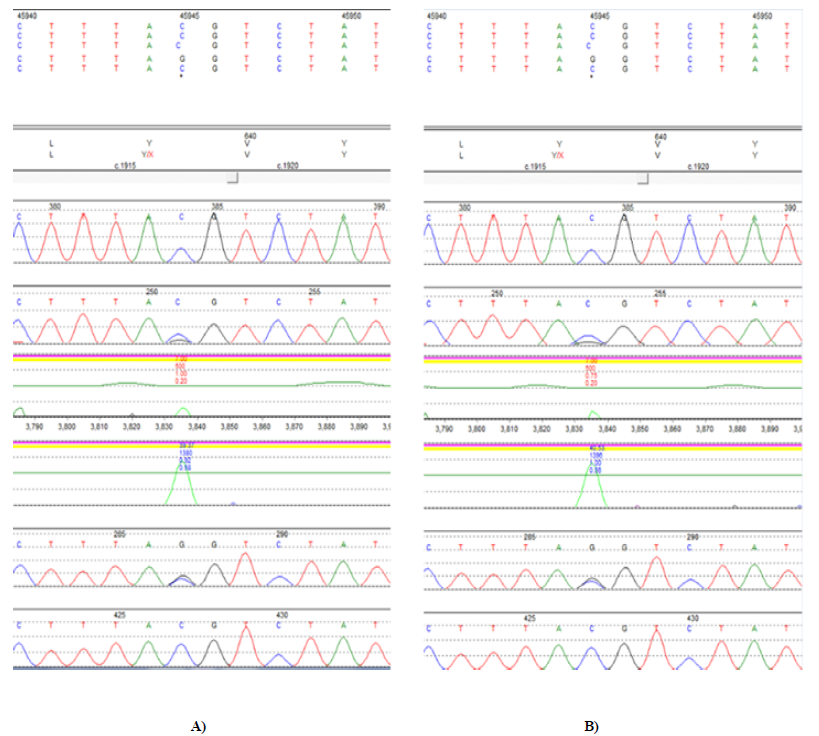
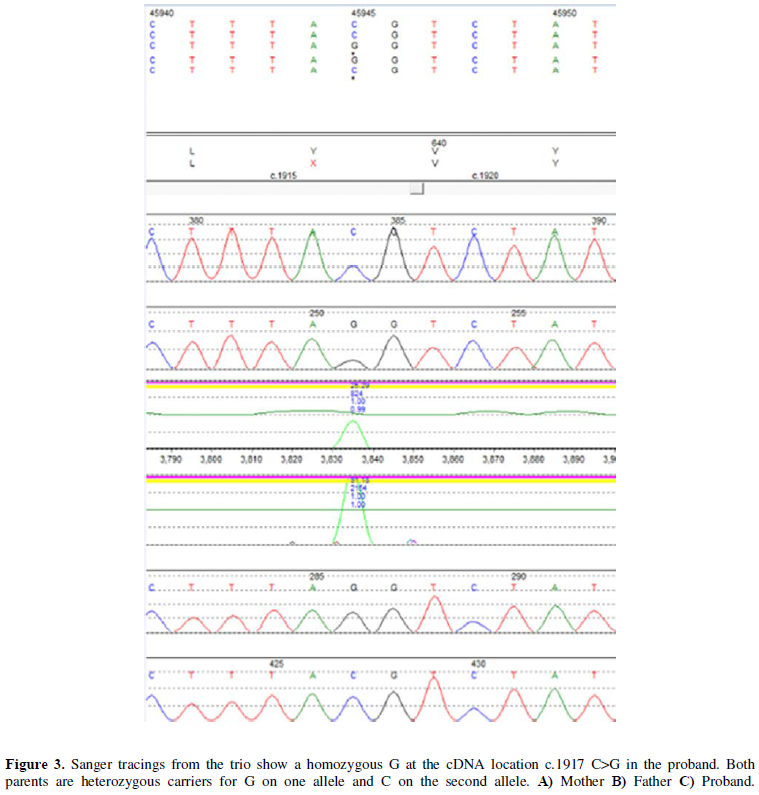
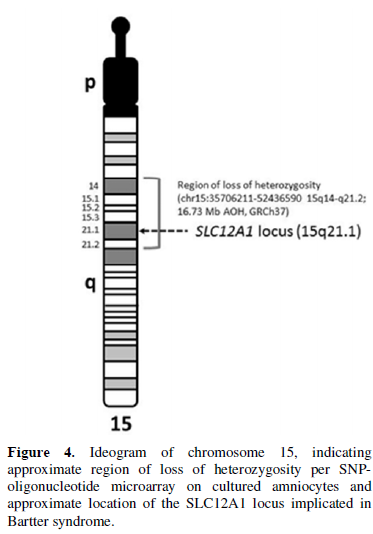
The infant presented to the hospital at 2 months of age with failure to thrive. The Pediatric Genetics service was consulted, as the etiology of the polyuria and electrolytes imbalances was not identified. Expedited whole exome sequencing revealed homozygosity for a truncating loss of function variant, c.1917C>G (p.Y639X) in the SLC12A1 gene (MIM 600839), consistent with the diagnosis of autosomal recessive Bartter Syndrome Type 1 (Figure 3). The region of chromosome 15q21.1 containing this gene was inherited from a common ancestor per microarray results (Figure 4). No other pathogenic, likely pathogenic, or variants of unknown significance were reported.
DISCUSSION
Bartter syndrome is a rare inherited renal tubular disorder associated with hypokalemic alkalosis. Hyperreninemic hyperaldosteronism with normal blood pressure, increased excretion of urinary prostaglandin E and hyperplasia of the juxtaglomerular apparatus are other features of this syndrome [19]. The antenatal form of this disorder is a severe condition that manifests as unexplained severe polyhydramnios with its associated complications of preterm labor and delivery, postnatal dehydration and electrolyte disturbances. It can lead to significant morbidity and mortality if undiagnosed and untreated, with some women considering termination of pregnancy. Due to the severity of this condition, we advocate prenatal diagnosis for prenatal and postnatal management.
There is inconsistency of amniotic fluid biochemistry and hormone levels in the diagnosis of Bartter syndrome [20-23]. In fact, controversial results of amniotic fluid electrolytes have been reported, especially elevated chloride levels in several case reports [24,25]. In contrast, a study of 16 cases of Bartter syndrome revealed no significant differences in amniotic fluid chloride levels [22]. Furthermore, elevated amniotic fluid aldosterone levels have also been described previously [20], while more recent evidence demonstrates that amniotic fluid aldosterone levels cannot be used for prenatal diagnosis of Bartter syndrome [26]. This case study highlights the difficulty of antenatal diagnosis of Bartter syndrome with traditional genetic methods and the opportunity to diagnose it prenatally using WES. Recent professional position statements recommend utilizing prenatal WES in cases of at least one major anomaly suggestive of a possible genetic etiology with a normal microarray. This case highlights the potential utility of WES in the setting of unexplained polyhydramnios with normal fetal anatomy and nondiagnostic microarray to aid in the diagnosis of antenatal Bartter Syndrome. Additionally, we highlight the usefulness of WES in cases where microarray identifies regions of homozygosity by descent indicating an increased risk for a single-gene recessive condition. Further studies examining the clinical utility and cost-benefit ratio of prenatal WES in cases such as ours are clearly needed before expanded recommendations for utilization can be made.
1. Bundgaard A, Andersen BR, Rode L, Lebech M, Tabor A (2007) Prevalence of polyhydramnios at a Danish hospital - A population-based study. Acta Obstet Gynecol Scand 86: 1427-1431.
2. Kollmann M, Voetsch J, Koidl C, Schest E, Haeusler M, et al. (2014) Etiology and perinatal outcome of polyhydramnios. Ultraschall Med 35: 350-356.
3. Magann EF, Chauhan SP, Doherty DA, Lutgendorf MA, Magann MI, et al. (2007) A review of idiopathic hydramnios and pregnancy outcomes. Obstet Gynecol Surv 62: 795-802.
4. Abele H, Starz S, Hoopmann M, Yazdi B, Rall K, et al. (2012) Idiopathic polyhydramnios and postnatal abnormalities. Fetal Diagn Ther 32: 251-255.
5. Wiegand SL, Beamon CJ, Chescheir NC, Stamilio D (2016) Idiopathic polyhydramnios: Severity and perinatal morbidity. Am J Perinatol 33: 658-664.
6. Bartter FC, Pronove P, Gill JR Jr, MacCardle RC (1962) Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am J Med 33: 811-828.
7. Jeck N, Schlingmann K, Komhoff M, Peters M, Waldegger S, et al. (2005) Salt handling in the distal nephron: Lessons learned from inherited human disorders. Am J Physiol Regul Integr Comp Physiol 288: 782-795.
8. Proesmans W (1997) Bartter syndrome and its neonatal variant. Eur J Pediatr 156: 669-679.
9. Ammenti, A, Montali S, (1996) "Neonatal variant" of Bartter syndrome presenting with acidosis. Pediatr Nephrol 10: 79-80.
10. McCredie DA, Rotenberg E, Williams AL (1974) Hypercalciuria in potassium-losing nephropathy: A variant of Bartter's syndrome. Aust Paediatr J 10: 286-295.
11. Hayward J, Chitty LS (2018) Beyond screening for chromosomal abnormalities: Advances in non-invasive diagnosis of single gene disorders and fetal exome sequencing. Semin Fetal Neonatal Med 23: 94-101.
12. Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, et al. (2013) Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med 369: 1502-1511.
13. Best S, Wou K, Vora N, Van der Veyver IB, Wapner R, et al. (2018) Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat Diagn 38: 10-19.
14. Mellis R, Chandler N, Chitty LS (2018) Next generation sequencing and the impact on prenatal diagnosis. Expert Rev Mol Diagn.
15. ACMG Board of Director (2013) Points to consider for informed consent for genome/exome sequencing. Genet Med 15: 748-749.
16. International Society of Prenatal Diagnosis, the Society of Maternal and Fetal Medicine and the Perinatal Quality Foundation (2018) Joint Position Statement from the International Society for Prenatal Diagnosis (ISPD), the Society for Maternal Fetal Medicine (SMFM) and the Perinatal Quality Foundation (PQF) on the use of genome-wide sequencing for fetal diagnosis. Prenat Diagn 38: 6-9.
17. Horn R, Parker M (2018) Opening Pandora's box?: Ethical issues in prenatal whole genome and exome sequencing. Prenat Diagn 38: 20-25.
18. Tayoun ANA, Spinner NB, Rehm HL, Green RC, Bianchi DW (2018) Prenatal DNA sequencing: Clinical, counseling and diagnostic laboratory considerations. Prenat Diagn 38: 26-32.
19. Phillips DR, Ahmad KI, Waller SJ, Meisner P, Karet FE (2006) A serum potassium level above 10 mmol/l in a patient predisposed to hypokalemia. Nat Clin Pract Nephrol 2: 340-346.
20. Nakanishi T, Suzumori N, Mizuno H, Suzuki K, Sato T, et al. (2005) Elevated aldosterone in amniotic fluid and maternal blood has diagnostic potential in pregnancies complicated with a fetus of Bartter syndrome. Fetal Diagn Ther 20: 481-484.
21. Matsushita Y, Suzuki Y, Oya N, Kajiura S, Okajima K, et al. (1999) Biochemical examination of mother's urine is useful for prenatal diagnosis of Bartter syndrome. Prenat Diagn 19: 671-673.
22. Garnier A, Dreux S, Vargas-Poussou R, Oury JF, Benachi A, et al. (2010) Bartter syndrome prenatal diagnosis based on amniotic fluid biochemical analysis. Pediatr Res 67: 300-303.
23. Massa G, Proesmans W, Devlieger H, Vandenberghe K, Van Assche A, et al. (1987) Electrolyte composition of the amniotic fluid in Bartter syndrome. Eur J Obstet Gynecol Reprod Biol 24: 335-340.
24. Proesmans W, Massa G, Vandenberghe K, Assche VA (1987) Prenatal diagnosis of Bartter syndrome. Lancet 1: 394.
25. Dane B, Yayla M, Dane C, Centin A (2007) Prenatal diagnosis of Bartter syndrome with biochemical examination of amniotic fluid: case report. Fetal Diagn Ther 22: 206-208.
26. Rachid M, Dreux S, Czerkiewicz I, Deschênes G, Muller F, et al. (2017) Prenatal diagnosis of Bartter syndrome: Amniotic fluid aldosterone. Ann Biol Clin 75: 204-208.
QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Carcinogenesis and Mutagenesis Research (ISSN: 2643-0541)
- International Journal of Radiography Imaging & Radiation Therapy (ISSN:2642-0392)
- Journal of Neurosurgery Imaging and Techniques (ISSN:2473-1943)
- Journal of Infectious Diseases and Research (ISSN: 2688-6537)
- Advance Research on Alzheimers and Parkinsons Disease
- Journal of Ageing and Restorative Medicine (ISSN:2637-7403)
- Journal of Rheumatology Research (ISSN:2641-6999)


