
648
Views & Citations10
Likes & Shares
This research focused on four crucial receptors of T. cruzi: 3GW9, 1ME3, 1SOI and 1AOG [8]. These receptors are essential for the parasite's survival and pathogenicity, making them prime targets for therapeutic intervention. 1ME3 is known for its role in the parasite's metabolic pathways, particularly in glycosomal processes, which are essential for energy production and the survival of T. cruzi. 1AOG is associated with the parasite's oxidative stress response mechanisms, making it a key target for disrupting the parasite’s ability to manage reactive oxygen species [9]. 1SOI is involved in the parasite's DNA replication and repair processes, crucial for maintaining the genetic integrity of T. cruzi. 3GW9 is linked to the parasite's cell signaling pathways, playing a significant role in its growth and development [10].
Ivan et al carried out a study on 32 Mannich base derivatives containing imidazole and benzimidazole as lead compounds for the treatment of T. cruzi. Mannich base represent promising alternatives to current anti-parasitic agents due to their potential biological activity against parasites responsible for tropical disease especially trypanosomiasis [11]. The aim of this research is to carry out virtual screening of derivatives against four protein receptors with PDB IDs: 3GW9, 1ME3, 1SOI and 1AOG as well as the pharmacokinetics evaluation of the derivatives with best binding affinities to serve as base line for Trypanosomiasis therapeutic agents. Studying these compounds is significant because of their potential to disrupt vital biological processes of T. cruzi, paving the way for novel treatment strategies (Table 1).
Table 1. Structures and binding affinities of the ligands with their target receptors.
|
S/N |
Compounds |
3GW9 |
1ME3 |
1SOI |
1AOG |
|
1 |
-16.9878 |
-18.482 |
-8.90891 |
-20.0865 |
|
|
2 |
-14.8951 |
-20.0245 |
-11.061 |
-20.4388 |
|
|
3 |
-17.9623 |
-18.7719 |
-9.34002 |
-18.2147 |
|
|
4 |
-22.4067 |
-20.188 |
-15.1585 |
-16.1542 |
|
|
5 |
-21.0917 |
-20.5135 |
-12.6872 |
-17.313 |
|
|
6 |
-16.3901 |
-21.1086 |
-9.86019 |
-20.0019 |
|
|
7 |
-19.6848 |
-19.6513 |
-13.4289 |
-16.2842 |
|
|
8 |
-18.3103 |
-20.3658 |
-5.23928 |
-22.3551 |
|
|
9 |
-16.6136 |
-20.1077 |
-18.1885 |
-16.387 |
|
|
10 |
-15.6946 |
-19.6985 |
-5.99739 |
-15.3737 |
|
|
11 |
-18.925 |
-20.7891 |
-13.4839 |
-18.3026 |
|
|
12 |
-19.2432 |
-19.7705 |
-5.73124 |
-21.4544 |
|
|
13 |
-20.2454 |
-22.2237 |
-14.9193 |
-19.2304 |
|
|
14 |
-17.5354 |
-20.659 |
-14.2092 |
-17.8839 |
|
|
15 |
-21.6673 |
-19.6782 |
-11.6228 |
-17.664 |
|
|
16 |
-16.1164 |
-21.9957 |
-13.3103 |
-16.4743 |
|
|
17 |
-18.0381 |
-18.2187 |
-20.4492 |
-17.945 |
|
|
18 |
-21.4017 |
-20.2127 |
-11.2788 |
-13.2617 |
|
|
19 |
-20.9439 |
-19.1622 |
-8.88809 |
-16.9156 |
|
|
20 |
-17.0932 |
-18.5937 |
-12.6202 |
-17.3412 |
|
|
21 |
-13.8364 |
-19.7172 |
-9.97625 |
-23.9657 |
|
|
22 |
-18.4112 |
-19.4641 |
-12.6133 |
-21.585 |
|
|
23 |
-17.612 |
-19.6439 |
-9.73277 |
-16.3431 |
|
|
24 |
-17.0044 |
-25.7809 |
-3.79164 |
-17.0073 |
|
|
25 |
-18.2856 |
-21.1711 |
-19.9064 |
-20.1567 |
|
|
26 |
-18.9996 |
-25.44 |
-6.56408 |
-15.5387 |
|
|
27 |
-18.6134 |
-24.9371 |
-3.0457 |
-17.4978 |
|
|
28 |
-23.5442 |
-21.4177 |
-8.37205 |
-17.0355 |
|
|
29 |
-19.3925 |
-19.5821 |
-6.33522 |
-21.603 |
|
|
30 |
-20.987 |
-22.2291 |
-15.6192 |
-10.7807 |
|
|
31 |
-15.0901 |
-17.8744 |
-7.18019 |
-8.4986 |
|
|
32 |
-22.1919 |
-20.5071 |
-16.182 |
-19.1199 |
MATERIALS AND METHODS
Data collection
The dataset used in this research consist of 32 series of Mannich base derivatives containing imidazole and benzimidazole as lead compounds for the treatment of T. cruzi collected from the literature of published work [4].
Sketching the structure
The obtained derivatives were sketched using Chem-Draw Ultra V12.0 software which is a comprehensive chemical drawing software developed by PerkinElmer [12], widely used in the field of chemistry for creating detailed chemical structures, reaction schemes, and molecular diagrams [13]. The software is user-friendly, allowing chemists and researchers to efficiently create and modify chemical structures, design reaction mechanisms, and visualize molecular interactions and save in CS file format [14].
Energy Minimization and Structure Optimization
The minimization of energy and geometry optimization is the process of finding the equilibrium or lowest energy geometry of molecules. The molecular geometry of all the structures were optimized with Spartan 14 V1.1.4 using B3LYP/6-311G*, the compounds were optimized using the density functional theory (DFT) with Becke‘s three-parameter hybrid functional [12] using LYP correlation functional [15]. The triple-zeta split-valence basis set (6-311G*) was used. The choice of DFT/B3LYP functional was due to the reason that the method leads to a satisfactory result when molecular geometries and energies are considered [6]. The result of optimizations was saved in SDF file format for docking analysis.
Molecular docking
The target protein structures were imported accordingly (usually in PDB format) as shown in Table 1, The protein feature was cleaned to remove any unwanted molecules such as water, ligands, or cofactors [17]. It was ensured that all hydrogen atoms were added, and the protonation states were also checked. The binding pocket were also identified.
The ligands were loaded as SDF file format to ICM-pro. The structures were optimized, ionization states and tautomeric forms were balanced using the ICM-pro software [17]. The receptor (protein) and ligands docked were selected, the grid box around the binding site was set, the docking algorithm and scoring function were chosen [18]. The docking process was initiated by clicking run docking or dock chemical tables. The docking process was monitored as ICM-Pro search for optimal binding poses by scoring them based on the selected algorithm [17].
Pharmacokinetics
The online tools PkCSM (<http://structure.bioc.cam.ac.uk/pkcsm>) and SwissADME (<http://www.swissadme.ch/index.php>) were utilized to assess the pharmacokinetic properties and drug-likeness of small molecules [19]. The online software was used to find topological polar surface area (TPSA) which is an indicative of membrane permeability, Log Kp (Skin Permeability) which measures skin permeability, gastrointestinal absorption, blood-brain barrier (BBB) permeability indicating potential activity in the central nervous system, CYP1A2 Inhibition which lead to drug-drug interactions if used concurrently with other medications [20].
RESULTS AND DISCUSSION
Docking Studies Result
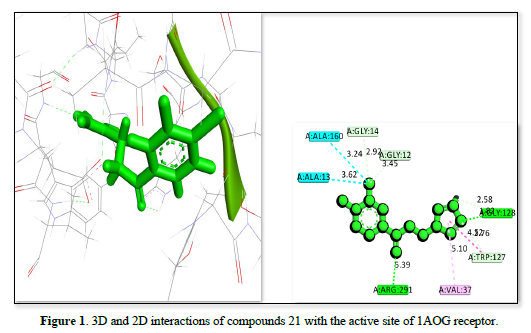
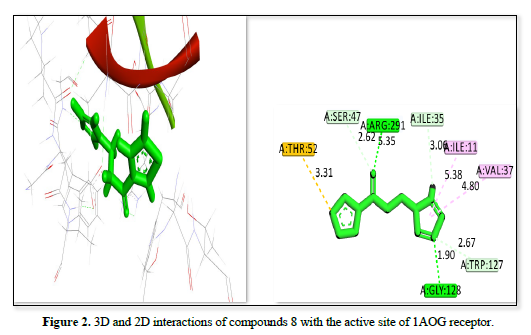
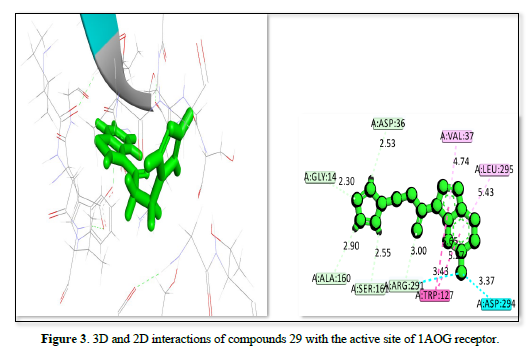
The docking analysis for three ligands as shown in Table 2 and Figures 1-3 interacting with the receptor (PDB ID: 1AOG) reveals varying degrees of binding affinity and interaction profiles. Ligand 21 exhibits the strongest binding affinity of -23.9657kj/mol, indicating a robust interaction with the receptor. It forms critical interactions with residues like ALA:160, GLY:14, ALA:13, and GLY:12 at bond distances ranging from 2.92 Å to 3.62 Å, which are essential for stabilizing the ligand within the binding pocket. Other interactions include GLY:128 and VAL:37, further contributing to the ligand's stability and binding efficacy. Ligand 8 showed a slightly lower binding affinity of -22.3551 but still demonstrates a strong interaction with the receptor. Key residues like ASP:36, GLY:14, ALA:160, and TRP :127 interacted at a shorter bond distances, ensuring the ligand's stability within the active site. Peripheral interactions with VAL :37, LEU:295, and ASP:294 help to further anchor the ligand, though they contribute less to the binding energy. Ligand 29 has the lowest binding affinity of -21.603, yet it still forms significant interactions with the receptor. Notable interactions include ASP:36, GLY:14, TRP :127, ARG :291, and SER:164, which stabilized the ligand within the active site. Peripheral residues like VAL:37 and LEU:295 also contribute to the overall stability but have less impact on the binding energy. All three ligands showed strong interactions with the receptor, with Ligand 21 having the highest binding affinity, followed by ligand 8 and ligand 29. The interactions involve key residues that stabilize the ligands within the receptor's binding site, supporting their potential as therapeutic agents.
Table 2. Receptor, Compound ID Amino acid residues, Bond distance and Binding affinity.
|
Receptor PDB ID |
Compound ID |
Amino Acid Residue |
Bond Distance (Å) |
Binding affinity (kj/mol) |
|
1AOG |
21 |
A: ALA:13 |
3.62 |
-23.9657 |
|
A: ALA:160 |
3.24 |
|||
|
A: GLY:14 |
2.92 |
|||
|
A: GLY:12 |
3.45 |
|||
|
A: ARG:291 |
5.39 |
|||
|
A: VAL:37 |
5.10 |
|||
|
A: TRP:127 |
4.52 |
|||
|
A: GLY:128 |
1.82 |
|||
|
1AOG |
8 |
A: THR:52 |
3.31 |
-22.3551 |
|
A: SER:47 |
2.62 |
|||
|
A: ARG:291 |
5.35 |
|||
|
A: ILE:35 |
3.0 |
|||
|
A: ILE:11 |
5.38 |
|||
|
A: VAL:37 |
4.80 |
|||
|
A: TRP:127 |
2.67 |
|||
|
A: GLY:128 |
1.90 |
|||
|
1AOG |
29 |
A: GLY:14 |
2.30 |
-21.6030 |
|
A: ASP:36 |
2.53 |
|||
|
A: VAL:37 |
4.74 |
|||
|
A: LEU:296 |
5.43 |
|||
|
A: ASP:294 |
3.37 |
|||
|
A: TRP:127 |
3.43 |
|||
|
A: ARG:291 |
3.00 |
|||
|
A: SER:164 |
2.55 |
|||
|
A: ALA:160 |
2.90 |
|||
|
1SOI |
17 |
A: GLU:230 |
2.87 |
-20.4492 |
|
A: ALA:59 |
3.93 |
|||
|
A: VAL:95 |
4.48 |
|||
|
A: ASP:96 |
3.76 |
|||
|
A: ARG:93 |
4.38 |
|||
|
A: ARG:53 |
1.68 |
|||
|
A: ARG:245 |
4.33 |
|||
|
A: ARG:314 |
6.47 |
|||
|
A: ARG:35 |
2.18 |
|||
|
1SOI |
25 |
A: ARG:93 |
2.40 |
-19.9064 |
|
A: VAL:95 |
3.72 |
|||
|
A: ALA:59 |
4.55 |
|||
|
A: SER:57 |
2.33 |
|||
|
A: ARG:58 |
6.19 |
|||
|
A: ASP:96 |
2.67 |
|||
|
A: TRP:120 |
5.64 |
|||
|
1SOI |
9 |
A: ARG:53 |
4.90 |
-18.8850 |
|
A: ARG:314 |
2.06 |
|||
|
A: ARG:35 |
6.02 |
|||
|
A: TYR:119 |
1.81 |
|||
|
A: ALA:59 |
4.58 |
|||
|
IME3 |
24 |
A: GLY:66 |
2.06 |
-25.7809 |
|
A: CYS:25 |
4.34 |
|||
|
A: LEU:67 |
4.00 |
|||
|
A: ALA:133 |
4.80 |
|||
|
A: GLU:205 |
2.54 |
|||
|
A: ASN:69 |
2.97 |
|||
|
A: MET:68 |
5.32 |
|||
|
IME3 |
26 |
A: LEU:67 |
4.40 |
-25.4400 |
|
A: CYS:25 |
5.50 |
|||
|
A: GLY:65 |
3.65 |
|||
|
A: GLY:66 |
1.72 |
|||
|
A: ALA:133 |
4.08 |
|||
|
A: MET:68 |
5.16 |
|||
|
IME3 |
27 |
A: CYS:25 |
5.78 |
-24.9371 |
|
A: LEU:67 |
5.42 |
|||
|
A: MET:68 |
4.41 |
|||
|
A: GLU:205 |
2.41 |
|||
|
A: ALA:133 |
4.64 |
|||
|
A: GLY:66 |
1.99 |
|||
|
3GW9 |
28 |
A: THR:296 |
1.95 |
-23.5442 |
|
A: GLY:414 |
2.78 |
|||
|
A: ILE:350 |
4.66 |
|||
|
A: PRO:355 |
4.82 |
|||
|
A: CYS:422 |
5.47 |
|||
|
A: ALA:291 |
4.88 |
|||
|
3GW9 |
4 |
A: ILE:350 |
4.71 |
-22.4067 |
|
A: PRO:355 |
4.97 |
|||
|
A: ALA:288 |
2.99 |
|||
|
A: ALA:291 |
3.96 |
|||
|
A: GLY:424 |
2.73 |
|||
|
A: THR:295 |
1.55 |
|||
|
3GW9 |
32 |
A: TRY:103 |
1.74 |
-22.1919 |
|
A: MET:358 |
2.06 |
|||
|
A: LEU:357 |
1.95 |
|||
|
A: LEU:354 |
2.89 |
|||
|
A: VAL:462 |
5.48 |
|||
|
A: LEU359 |
5.27 |
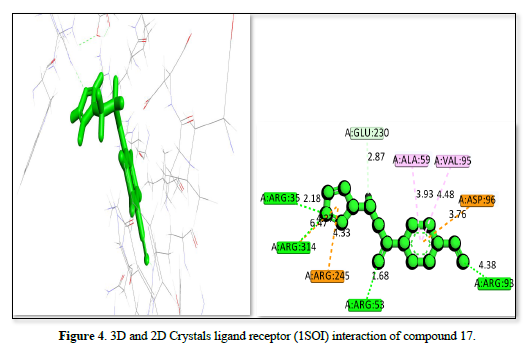
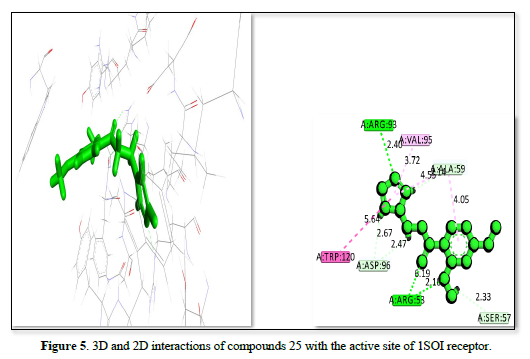
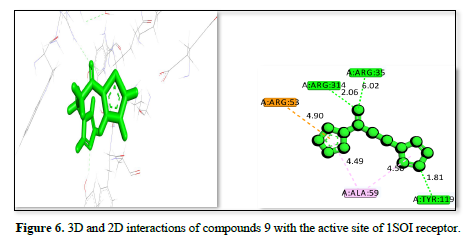
The complex of receptor and ligand shown in Table 2 and Figures 4-6 has a binding affinity of -20.4492kj/mol, -19.9064kj/mol and -18.885kj/mol, respectively. The interaction residues include GLU :230, ALA :59, VAL: 95, ASP :96, ARG :93, A: ARG :53, A: ARG :245, A: ARG:314, and ARG :35. The bond distance range from 1.68 Å (ARG A:53) to 6.47 Å (ARG A:314) for the Figure 4 complex, indicating a strong binding primarily through hydrogen bonds with ARG residues and hydrophobic interactions with other residues. The Figure 5 receptor ligand interacts with ARG :93, VAL A:95, ALA:59, SER:57, ARG:58, ASP:96, and A: TRP:120. Bond distance range from 2.33 Å (SER A:57) to 6.19 Å (ARG A:58), suggesting a balance of hydrogen bonds and hydrophobic interactions, with SER and TRP playing key roles in stabilizing the ligand. This ligand interacts with ARG :53, ARG :314, ARG:35, TYR:119, and ALA :59 amino acid residues. The bond distances range from 1.81 Å (ARG A:35) to 6.02 Å (ARG:314), showing a strong interaction with ARG residues and a significant contribution from TYR. The complex in figure 4 exhibits the strongest binding affinity due to its effective interactions with multiple ARG residues and balanced hydrogen bonding and hydrophobic interactions, also Figure 5 shows strong binding with a balance of interactions, while Figure 6, though with slightly lower affinity, still demonstrates significant interactions with key residues.
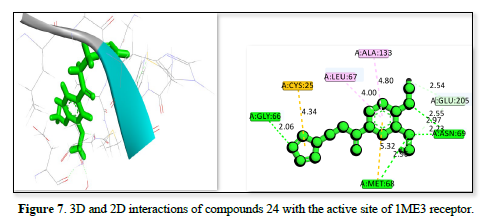
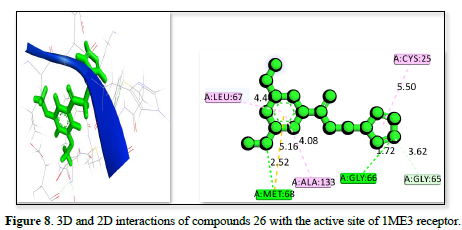
The studies of the IME3 receptor and its interaction with three ligands in Table 2 reveals strong binding affinities of -25.7809kj/ml, -25.44kj/mol, and -24.9371kj/mol respectively. All ligands form significant hydrogen bonds and hydrophobic contacts, with the strongest interactions involving residues like GLY:66, GLU :205, CYS :25, and MET :68 which has a bond distance ranging from 2.06 to 5.32 Å. The figure 7 demonstrates the strongest binding affinity due to optimal hydrogen bonding and polar interactions and has a bond distance ranging from 1.72 to 5.50Å, followed closely by Figures 8 & 9, which has a bond distance ranging from 1.99 to 5.78Å. These findings suggested that all three ligands have potential as potent inhibitors of the IME3 receptor.
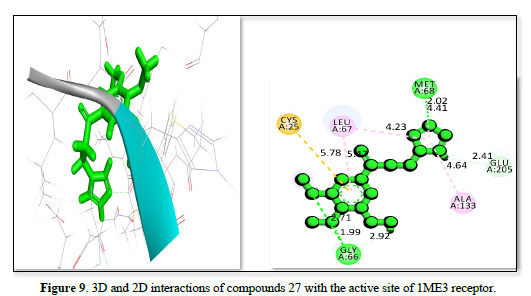
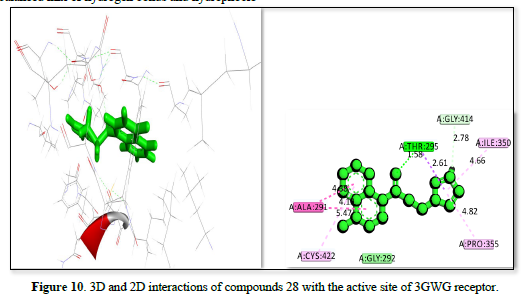
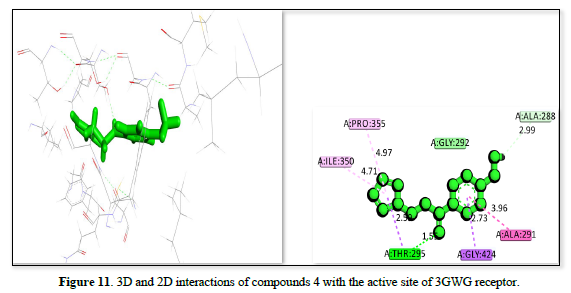
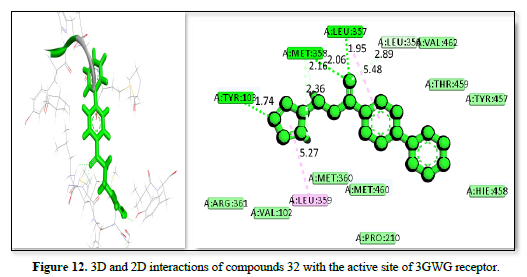
The study of 3GW9 receptor with ligands shown in Table 2 and Figures 10-12 shows the strongest binding affinity of -23.5442kj/mol, -22.4067kj/mol and -22.1919kj/mol respectively, and interacting closely with residues THR A:296, GLY A:414, ILE A:350, PRO A:355, CYS A:422, and ALA A:291 and a bond distance ranging from 1.98 to 5.47Å The strong hydrogen bonding with THR and GLY, combined with hydrophobic interactions with CYS and PRO, contributes to its high binding affinity. The complex displayed in Figure 11 interacts with amino acid residues ILE :350, PRO :355, ALA :288, ALA :291, GLY :424, and THR :295 and has a bond distance of 1.55 to 4.97Å. It exhibits a balanced mix of hydrogen bonds and hydrophobic interactions, with THR A:295 providing a strong polar interaction. The complex shown in Figure 12 interact with residues TRY :103, MET :358, LEU :357, LEU :354, VAL :462, and LEU :359 at a bond distance of 1.74 to 5.27Å. It has strong hydrogen bonding with TRY :103, but longer interaction distances with other residues reduce its overall binding strength compared to figure 10 complexes. The interaction in Figure 10 is the most promising candidate due to its strong binding interactions and effective hydrogen bonding with key residues. The interaction in Figures 11 & 12 also show good binding but with slightly weaker affinities due to longer interaction distances.
Pharmacokinetics studies
The pharmacokinetic studies for three compounds (21, 8, and 29) docked against 1AOG receptor were analyzed to evaluate their potential as therapeutic agents. The most essential parameters considered include: The molecular weight (MW), and this is important because a lower molecular weight generally leads to better absorption and distribution. Compound 8 has the lowest molecular weight (206.26 g/mol), while compound 29 has the highest (274.31 g/mol). Topological Polar Surface Area (TPSA) is indicative of membrane permeability. Compound 21, with a TPSA of 34.89 Ų, is likely to have superior permeability compared to compounds 8 and 29, which both have a TPSA of 63.13 Ų. Log Kp (Skin Permeability) measures skin permeability, and compound 8 has the lowest value (-6.89 cm/s), suggesting it is the least permeable through the skin. Compounds 21 and 29 show slightly better skin permeability. Gastrointestinal (GI) Absorption is high for all three compounds, indicating that they are likely to be well absorbed when taken orally. Blood-Brain Barrier (BBB) Permeability is present in each of the compounds, indicating potential activity in the central nervous system. CYP1A2 Inhibition is observed in all compounds, which could lead to drug-drug interactions if used concurrently with other medications metabolized by an enzyme (Table 3).
Table 3. Pharmacokinetics of the best docked ligands and their status.
|
Receptor: 1AOG |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ID |
MW |
TPSA |
XLOGP3 |
WLOGP |
MLOGP |
GIA |
BBBP |
CYP1A2 I |
log Kp (cm/s) |
BAS |
AMES-T |
hERG I |
ORCT |
Hep. T |
|
21 |
252.67 |
34.89 |
1.99 |
3.37 |
1.91 |
High |
Yes |
Yes |
-6.43 |
0.55 |
Yes |
No |
1.376 |
No |
|
8 |
206.26 |
63.13 |
0.94 |
2.22 |
0.44 |
High |
Yes |
Yes |
-6.89 |
0.55 |
Yes |
No |
1.815 |
Yes |
|
29 |
274.31 |
63.13 |
2.38 |
3.93 |
2.02 |
High |
Yes |
Yes |
-6.28 |
0.55 |
Yes |
No |
1.483 |
No |
|
Receptor: 1SOI |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ID |
MW |
TPSA |
XLOGP3 |
WLOGP |
MLOGP |
GIA |
BBBP |
CYP1A2 I |
log Kp (cm/s) |
BAS |
AMES-T |
hERG I |
ORCT |
Hep. T |
|
17 |
225.25 |
58.68 |
0.98 |
2.03 |
0.31 |
High |
Yes |
Yes |
-6.98 |
0.55 |
Yes |
No |
2.448 |
No |
|
25 |
260.29 |
53.35 |
1.2 |
2.17 |
0.36 |
High |
Yes |
Yes |
-7.04 |
0.55 |
Yes |
No |
1.288 |
Yes |
|
9 |
206.26 |
63.13 |
1.27 |
2.22 |
0.44 |
High |
Yes |
Yes |
-6.66 |
0.55 |
Yes |
No |
1.815 |
Yes |
|
Receptor: IME3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ID |
MW |
TPSA |
XLOGP3 |
WLOGP |
MLOGP |
GIA |
BBBP |
CYP1A2 I |
log Kp (cm/s) |
BAS |
AMES-T |
hERG I |
ORCT |
Hep. T |
|
24 |
260.29 |
53.35 |
1.2 |
2.17 |
0.36 |
High |
Yes |
Yes |
-7.04 |
0.55 |
Yes |
No |
1.143 |
No |
|
26 |
260.29 |
53.35 |
1.2 |
2.17 |
0.36 |
High |
Yes |
Yes |
-7.04 |
0.55 |
Yes |
No |
1.109 |
Yes |
|
27 |
290.31 |
62.58 |
1.17 |
2.18 |
0.07 |
High |
Yes |
Yes |
-7.24 |
0.55 |
No |
No |
1.147 |
No |
|
Receptor: 3GW9 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ID |
MW |
TPSA |
XLOGP3 |
WLOGP |
MLOGP |
GIA |
BBBP |
CYP1A2 I |
log Kp (cm/s) |
BAS |
AMES-T |
hERG I |
ORCT |
Hep. T |
|
28 |
268.29 |
34.89 |
2.61 |
3.87 |
2.21 |
High |
Yes |
Yes |
-6.08 |
0.55 |
Yes |
No |
1.785 |
No |
|
4 |
230.26 |
44.12 |
1.23 |
2.16 |
0.66 |
High |
Yes |
Yes |
-6.83 |
0.55 |
Yes |
No |
1.819 |
No |
|
32 |
276.33 |
34.89 |
2.89 |
3.82 |
2.26 |
High |
Yes |
Yes |
-5.93 |
0.55 |
No |
No |
1.875 |
No |
KEY: ID: Compound; MW: Molecular Weight; TPSA: Topological Polar Surface Area; XLOGP3, WLOGP, MLOGP: Lipophilicity Values; GIA: Gastrointestinal Absorption; BBBP: Blood-Brain Barrier Permeability; CYP1A2 I: Cytochromes Inhibitors; BAS: Bioavailability Score; AMES--Toxicity ORCT: Oral Rat Chronic Toxicity; hERG I: Toxicity Inhibitor; Hep. T: Hepatotoxicity
Table 3 show ligands with IDs 21, 8, and 29 and their pharmacokinetics properties. Their molecular weights are all within the permissible range for drug-likeness (usually < 500 g/mol). Ligand 21 has a lower TPSA than ligand 8 and ligand 29, which enhanced cell membrane permeability. TPSA values below 140Ų indicates good permeability. All three compounds have reasonable lipophilicity based on the lipophilicity values (XLOGP3, WLOGP, and MLOGP), with ligands 29 having the highest lipophilicity. Each molecule has a high GIA and has blood-brain barrier (BBBP = Yes). These findings suggest that all compounds have pass the blood-brain barrier and are well-absorbed orally, which is a desired characteristic for medications that act on the central nervous system. All exhibit variable skin permeability (log Kp values: -6.43, -6.89, and -6.28 cm/s) and has a potential sign of drug-drug interactions CYP1A2 inhibition. ligand 29 have the maximum permeability, the low log Kp values indicate moderate skin permeability. AS = 0.55 for all substances stipulate a moderate oral bioavailability. AMES test results show that they are mutagenic (Yes). The hERG inhibition value of negative (No) for all ligands indicating no cardiac toxicity risk. The Oral rat chronic toxicity (ORCT) for ligand 8 is positive, stipulating long-term toxicity. However, Ligands 21 and ligand 29 are hepatotoxicity (Hep. T = No) showing oral rat chronic toxicity (ORCT = No).
Three ligands were analyzed with IDs 17, 25, and 9 shown in Table 3 and the results of their molecular weights is within the usual range for drug-likeness (below 500 g/mol). The TPSA values are less than 140Ų, showing good permeability through cell membranes. The moderate lipophilicity values for all three molecules enhance the medication solubility and absorption showing a well-balanced property, these findings imply that all three substances have pass across the blood-brain barrier and can be absorbed when taken orally. This characteristic is important for medications that target the central nervous system (CNS). Since CYP1A2 is involved in the metabolism of numerous medicines, inhibition of CYP1A2 showcased the possibility of drug-drug interactions. All have limited skin permeability based on the negative log Kp values. Ligands have a Bioavailability Score (BAS) of 0.55, oral bioavailability is moderate. The AMES test results are positive (Yes), hERG inhibition negative (No), Ligand 17 and 25 as well as ligand 9.
Three compounds with IDs 24, 26, and 27, have shown promising qualities for therapeutic development. The properties that all three drugs have in common are high gastrointestinal absorption (GIA), oral bioavailability. blood-brain barrier (BBBP = Yes), Similar XLOGP3, WLOGP, and MLOGP values demonstrate the moderate lipophilicity of each chemical, guaranteeing that hydrophobicity and hydrophilicity are successfully balanced. These characteristics proved their distribution and solubility in biological systems. All three compounds were predicted to inhibit CYP1A2, pointing to a potential for drug-drug interactions during metabolism. Their skin permeability, measured by log Kp values, is relatively low, indicating that these compounds are less effective in transdermal applications. Despite the similarities in their absorption and distribution profiles, their toxicity assessments present key differences. As shown in Table 3.
CONCLUSION
Finally, the complex binding affinity obtained range from -25.7809kj/mol to -18.885 when the four receptors were docked against the derivatives so far, they are better when compared to the approved Drug NIFURTIMOX which has the binding affinity of -14.445kj/mol. The standard Drug in terms of ADME and Pharmacokinetics analysis is less effective because it does not meet up some of the standard parameters set up for Drug candidate. However, the advancement of computational chemistry tool enhances the improvement of the therapeutics drug and validation of some existing drug for better treatment and reduce damages caused. The values of the topological polar surface area (TPSA) and oral rat chronic toxicity (ORCT) of the best derivatives are all within the accepted range but that of the standard (NIFURTIMOX) have 117.08Ų and 1.868mg/kg, showing that they do not concord with the accepted parameters. The blood-brain barrier of the of the standard (No) showing that substances cannot pass across the blood-brain barrier properly and cannot be absorbed well when taken orally, this may be the reason that leads to higher toxicity when NIFURTIMOX is administered for a long time. The complexes as compared to the standard (NIFURTIMOX) have better drug interaction meaning it can inhibit CYP1A2, pointing to a potential for drug-drug interactions during metabolism.
- Lidani KC, Andrade FA, Bavia L, Damasceno FS, Beltrame MH, et al. (2019) Chagas disease: from discovery to a worldwide health problem. Front Public Health 7: 166.
- Rocha-Ortega M, Nava-Bolaños A, Córdoba-Aguilar A (2024) Merging socioecological variables to predict risk of Chagas disease. Acta Tropica 251: 107098.
- Stijlemans B, De Baetselier P, Van Molle I, Lecordier L, Hendrickx E, et al. (2024) Q586B2 is a crucial virulence factor during the early stages of Trypanosoma brucei infection that is conserved amongst trypanosomatids. Nat Commun 15(1): 1779.
- World Health Organization (2021) WHO fact sheets, Chagas disease.
- Franco JR, Cecchi G, Priotto G, Paone M, Ebeja AK, et al. (2022) Human African trypanosomiasis cases diagnosed in non-endemic countries (2011-2020). PLoS Negl Trop Dis 16(11): e0010885.
- Bhattacharya A, Corbeil A, do Monte-Neto RL, Fernandez-Prada C (2020) Of drugs and trypanosomatids: New tools and knowledge to reduce bottlenecks in drug discovery. Genes 11(7): 722.
- Tefe-Silva C, Teixeira LD, Durigan LR, Cardoso MC, Davi ML, et al. (2023) The Role of MicroRNAs in the Pathogenesis of Chagas Disease. Int J Cardiovasc Sci 36: e20220210.
- Gomes FM, Silva M, Molina-Cruz A, Barillas-Mury C (2022) Molecular mechanisms of insect immune memory and pathogen transmission. PLoS Pathog 18(12): e1010939.
- Bern C (2015) Chagas’ disease. N Engl J Med 373(5): 456-466.
- Beltran-Hortelano I, Atherton RL, Rubio-Hernández M, Sanz-Serrano J, Alcolea V, et al. (2021) Design and synthesis of Mannich base-type derivatives containing imidazole and benzimidazole as lead compounds for drug discovery in Chagas Disease. Eur J Med Chem 223: 113646.
- Conners EE, Vinetz JM, Weeks JR, Brouwer KC (2016) A global systematic review of Chagas disease prevalence among migrants. Acta Tropica 156: 68-78.
- Isa A, Uzairu A, Umar U, Ibrahim MT, Umar A (2024) QSAR, docking and pharmacokinetic studies 2, 4-diphenyl indenol [1, 2-B] pyridinol derivatives targeting breast cancer receptors. J Chem Lett 5: 44-57.
- Kubo AI, Uzairu A, Babalola IT, Ibrahim MT, Umar AB (2024) QSAR, molecular docking, and pharmacokinetic analysis of thiosemicarbazone-indole compounds targeting prostate cancer cells. J Taibah Univ Med Sci 19(4): 823-834.
- Ibrahim MT, Uzairu A, Shallangwa GA, Ibrahim A (2020) In-silico studies of some oxadiazoles derivatives as anti-diabetic compounds. J King Saud Univ Sci 32(1): 423-432.
- Abdulfatai U, Uzairu A, Uba S (2017) Quantitative structure-activity relationship and molecular docking studies of a series of quinazolinonyl analogues as inhibitors of gamma amino butyric acid aminotransferase. J Adv Res 8(1): 33-43.
- Adedirin O, Uzairu A, Shallangwa GA, Abechi SE (2018) QSAR and molecular docking-based design of some n-benzylacetamide as? -aminobutyrate-aminotransferase inhibitors. J Eng Exact Sci 4(1): 65-84.
- Adawara SN, Shallangwa GA, Mamza PA, Ibrahim A (2020) Molecular docking and QSAR theoretical model for prediction of phthalazinone derivatives as new class of potent dengue virus inhibitors. Beni-Suef Univ J Basic Appl Sci 9: 1-7.
- Adawara SN, Shallangwa GA, Mamza PA, Ibrahim A (2021) In-silico approaches towards the profiling of some anti-dengue virus as potent inhibitors against dengue NS-5 receptor. Sci Afr 13: e00907.
- Ibrahim MT, Tabti K, Abdulsalam S, Tahir AS, Mahmoud A, et al. (2024) Discovery of new 2, 4-diaminopyrimidines derivatives as EGFRT790M kinase inhibitors: A structure-based approach with DFT calculation, drug-likeness, ADME-toxicity properties evaluation and MD simulation. J Umm Al-Qura Univ Appl Sci 10: 257-273.
- Ibrahim MT, Uzairu A, Shallangwa GA, Uba S (2020) Computer-aided molecular modeling studies of some 2, 3-dihydro-[1, 4] dioxino [2, 3-f] quinazoline derivatives as EGFR WT inhibitors. Beni-Suef Univ J Basic Appl Sci 9: 1-0.














