
1007
Views & Citations7
Likes & Shares
The influence of water molecules in different positions on the dye 4-[[(2,4-dinitrophenyl)methylene]imino-2,6-diphenyl]phenol is studied by the quantum chemical calculations using the method CAM-B3LYP and the basis sets 6-31+G(d) and 6-311+G(d,p). In general, the phenol fragment (position 1) is hydrated first as it is seen from the fact that a hydrogen atom in the neutral (protonated) form attaches to it. However, hydration of the dinitrophenol fragment (position 2) has a greater influence on the absorption wavelength. In pure water, the dye is present in the form of an anion hydrated by 5 water molecules: NPh-+1H2O(1)+ 4H2O(2), 1 molecule in position 1 and 4 molecules in position 2. Solvation shells of the Reichardt’s dye and 4-[[(2,4-dinitrophenyl)methylene]imino-2,6-diphenyl]phenol in water are also studied by the molecular dynamics simulation. A range of specific interactions is detected, and it is noted that the protonated and deprotonated models for the dye 4-[[(2,4-dinitrophenyl)methylene]imino-2,6-diphenyl]phenol retain water in the first solvation shell better than the Reichardt’s dye. These results lead to the conclusion that the dye 4-[[(2,4-dinitrophenyl)methylene]imino-2,6-diphenyl]phenol is more informative in comparison with the Reichardt’s dye in the study of mixed water-based solutions.
Keywords: Solvatochromy, Solvatochromic dye, Solvent polarity, Quantum chemical calculation, Polarizable continuum model (PCM), Molecular dynamics simulation, Solvation shell

Due to this, scientists do not still give up hope to synthesize new dyes, which could be more informative in this and other issues. [3-9] One of such dyes is 4-[[(2,4-dinitrophenyl)methylene]imino-2,6-diphenyl]phenol (hereinafter Ind 2) (Figure 2) [9]. Ind 2 has 2 solvation centers that change depending on the composition of the mixed water-organic solution. A more detailed difference between the molecular shells of Ind 1 and Ind 2 dyes was previously studied by our group [10].

QUANTUM CHEMICAL CALCULATIONS
The influence of the solvation of a molecule of the dye Ind 2 on the absorption wavelength is studied by the method of quantum chemical calculations. A similar study for Ind 1 has been earlier carried out by a group of scientists from Belgium and France [11]. Calculations are made in the Gaussian packet [12] using the CAM-B3LYP method and the basis sets 6-31+G(d) and 6-311+G(d,p). To calculate the energy of electronic transitions, the basis set must contain polarization and diffusion orbitals. Also, calculations for a large number of configurations of big molecular aggregates are needed. This is a reason for choosing the least admissible basis set. First of all, electronic configuration of the ground state of the “dye – solution” molecular aggregate is calculated. At this, we add one or several water molecules so that they create hydrogen bonds with certain atoms of the dye molecule. This aggregate is considered to be surrounded by structureless solvent, the interaction with which is considered by the PCM (polarizable continuum model) method. The parameters ε and ε0 taken from literature data are specified for this. Thus, the quite strong specific influence of solvent molecules located near the dye and nonspecific long-range solvation weakly dependent on the solvent are considered. For the calculations used in this article, we optimize the geometry of the molecule, considering the influence of the solvent by the PCM method.
In the PCM method, uneven charge distribution inside the molecule polarizes dielectric continuum surrounding the molecule, for which the electric charge distribution is calculated. The polarized solvent, in its turn, creates the so-called reactive field. In the ground state, the reactive field polarizes electrons and bonds of molecules and reduces the total energy. In the excited state, orientation of the solvent molecules does not change, since the photon absorption process occurs very quickly. Thus, the reactive field for the calculation of the electronic configuration of the ground and excited states remains unchanged. At the second stage, the energy of excitement by the TDDFT procedure is calculated. The reactive field calculated at the first stage is used to consider the influence of the structureless solvent. Optimization of the geometry of the molecule in the excited state is not carried out, since the positions of atoms do not have time to change at the absorption of a photon according to the Franck-Kondon principle. The analysis of the charge structure and distribution in the molecular ion NPh- of the dye Ind 2 allows distinguishing of three centers, which can be significantly influenced by the solvent (Figure 3). Since the dye is an anion and has no acidic hydrogen atoms, such centers are hydrogen bond acceptors.
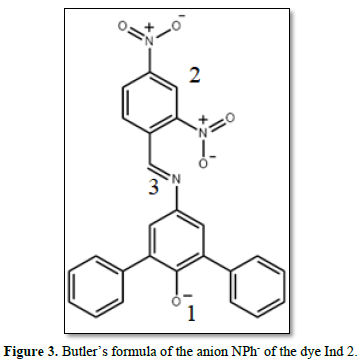
Center No. 1: a negatively charged oxygen atom has the greatest proton affinity. It is this atom that is protonated with the creation of a neutral molecule NPhH, which is the main form of the existence of the dye in its pure form (crystalline substance). Solutions of neutral molecules NPhH do not absorb light in the visible area.
Center No. 2: oxygen atoms of the dinitrophenyl fragment. In fact, these are as many as 4 centers, which can create 4 hydrogen bonds with solvent molecules.
Center No. 3: a nitrogen atom connecting two parts of the molecule. As it can be seen from Figure 4, it is in the state of sp2 hybridization and has a lone electronic pair, which can create the hydrogen bond.

Solvent molecules are located manually so that to create hydrogen bonds with coordination centers (Figure 4). The next stage is optimization of the geometry of received molecular aggregates in the ground state. At this, nonspecific solvation is considered using the PCM method. The results of the quantum chemical calculations are given in Table 1.

Hydration in position 1 decreases the absorption wavelength (667.13 => 599.12; ×0.898 for a water molecule), and protonation decreases it a lot (to 385.7 nm) (Table 2), which corresponds to experimental observations. Hydration in position 2 increases the absorption wavelength (667.13 => 695.49; ×1.043 for a water molecule).
If multiply the corrections for hydration in position 1 and position 2, we can receive a correction for the dye hydrated by two water molecules:

which is close to the estimated meaning of the system NPh− + 2H2O (1,2) of the value 621.53 nm calculated by the quantum chemical method.

The system NPh− without the PCM (in vacuum) is compared with experimental data of the studied dye in the non-polar solvent-hexane. The experimental value 621.54 nm is larger than the calculated one (574.36 nm) by 1.082 times (equation 3):

This factor can be used as an empirical correction to the calculated wavelengths, i.e., proceeding from this calculation, QCC(water) must be:

The configuration NPh− + 1H2O (2) has the closest value-695.49 nm. However, hydration of the dye in position 2 without hydration in position 1 turns out unlikely. This correction together with the assessment of the influence of hydration with a water molecule allows calculation of the absorption wavelength for different hydration options. At this, we assume that position 1 will be hydrated, since this oxygen atom has the most negative charge and is protonated in the acidic environment.


It can be seen from these calculations that the system NPh−+1H2O(1)+ 4H2O(2) has a result close to experimental data (equation 9). After these assumptions, additional calculations were carried out using the basis set 6-311+G(d,p) (Table 1). The third provision was not investigated. As you can see, those systems that were retested have slightly lower results compared to the basis set 6-31+G(d). The system NPh− + 5H2O (1,2,2,2,2) has a rather good result, which may correspond to reality, because it is quite difficult to achieve an exact reproduction of experimental results.
The received results demonstrate that the addition of water in the second position has the largest influence on the solvation of the dye Ind 2-it is well seen in the systems with the addition by this position only and with at least partial presence of water molecules by this position. i.e., we can observe specific solvation due to the presence of nitrogen-containing fragment. For the final verification of our hypothesis about which solvation center of Ind 2 is more prone to interaction with water, this system is also studied with the addition on a hydrogen atom in the first and second positions (the results are given in Figure 5 and in Table 2). The third position is not studied in this case.

The results show that a hydrogen atom in the configuration NPh− + 1H (2) does not almost influence the system, since if compare it to the system without water, namely NPh− with λ= 667.13 nm, we will notice a difference only in 1.77 % or 11.72 nm, i.e., the system does not undergo large changes. On the contrary, protonation in the first position (NPh− + 1H (1)) decreases the absorption wavelength a lot (by 42.19 %), which makes the molecule colorless. Also, the results of the molecular chemical simulation show that position 3 of the dye is not actually hydrated (less than 0.5 molecules are added in average), that is why we do not discuss the influence of hydration in this position on the absorption spectra. The absence of hydration can be explained by the effective charge of a hydrogen atom close to zero and its surrounding by large fragments of the molecule. In general, the phenol fragment (position 1) is hydrated first, which is seen from the fact that a hydrogen atom in the neutral (protonated) form is added to it. However, the hydration of the dinitrophenol fragment (position 2) has a larger influence on the absorption wavelength. In pure water, the dye is present in the form of an anion hydrated by 5 water molecules: NPh−+1H2O(1)+ 4H2O(2), 1 molecule in position 1 and 4 molecules in position 2.
MOLECULAR DYNAMICS SIMULATION
Calculations are also made to identify specific interactions, namely, hydrogen bonds between the molecules of the dyes (Ind 1 and Ind 2) and water by the molecular dynamics’ simulation using the software package MDNAES [13]. All-atom non-rigid models of the molecule Ind 2 (protonated (NPhH) and deprotonated (NPh) models) capable of reproducing the main conformations of this molecule are developed and validated in the course of the work. The models are created using the builder LigParGen providing the force field parameters OPLS-AA for organic molecules or ligands offered by the Jorgensen group [14-18]. Ind 1 is used for comparison. Its models are borrowed [19] (protonated (RDH) and deprotonated (RD) molecules are used). These models are calculated by the builder R.E.D. Server [20,21]. Total system energy is calculated as a sum of intramolecular energy and intermolecular energy:

Intramolecular energy is a sum of all intermolecular interaction potentials such as:

There are no intramolecular interactions 1-4.
In its turn, intermolecular energy is a sum of all intermolecular interaction potential, namely:

The Lorenz-Berthelot combining rule is used to calculate the Lennard-Jones potential. The reactive field method is used to calculate the Coulomb potential. Parameters of intramolecular bond potentials, parameters of intermolecular interaction and coordinates of atoms of the proposed models are given in the appendix. The rigid triatomic model of water-SPC/E is also used, in addition to the models of dyes [22].
Modeling details:
- Ensemble-NVT
- The numbers of water molecules-for Ind 2 - 255 molecules, for Ind 1 - 499 molecules
- Number of dye molecules-1 molecule
- ε = 78.3
- ρ = 997.19 kg/m3 (density of the entire system)
- T = 298.15 K
- Δt = 0.0005 ps
- Equilibration time-200,000 steps (100 ps)
- Calculation time-100,000 steps (50 ps).
During simulation of the solvation shell of the dyes, a water molecule is considered belonging to the solvation shell if the distance between the coordination centers of the dye and water does not exceed a certain value. The maximum distance between the coordination centers of the dye to water is taken from the first minimum on the radial distribution function (RDF).
Four calculations for each model are made to verify the location of water molecules in the systems:
- Two calculations provided the coordination center in a water molecule is hydrogen atoms, and in the dye molecule-all heavy atoms-C, O, N (Table 3, column 3, contains the results showing the number of water molecules in the system near the first and second positions (for Ind 1, the phenol fragment as the first position and the π-system as the second position are meant)).
- Two calculations provided the coordination center in a water molecule is an oxygen atom, and in the dye molecule-all hydrogen atoms (Table 3, column 5, contains the results showing the number of water molecules in the system near the first and second positions).
Table 3 (columns 4 and 6) also shows average numbers of water molecules located near the molecules of the dye during the calculations made (Figures 6-13).









The following hydrogen bonds are found according to the results of this study:
- Between water molecules with the coordination centers Н and О atoms of both dyes
- Between water molecules with the coordination centers Н and π-systems of both dyes
- Between water molecules with the coordination centers O and H atoms of both dyes.
Also, a specific hydrogen bond is found-between the water molecule and the atom N (in the third position) of the model NPhH, which is absent in the Reichardt’s dye, probably because of the large loading of the molecule. On the other hand, this bond cannot affect the solvation, since the studied dyes “work” only in the deprotonated state, and no similar bonds are found in the deprotonated models.
Based on the results of simulation, we can state that both models of Ind 2 attract a larger number of water molecules into their solvation shells than the similar models of Ind 1. Also, proceeding from the results of simulation, we can confirm our conclusion about the results of the quantum chemical calculations that in Ind 2, 2 fragments participate in the hydration process during interaction with water-nitrogen-containing and phenol ones. It should be also stated that the molecular dynamics simulation also confirms the assumption that for Ind 2, position 3 does not influence the solvation, since we can observe the full absence of hydrogen bonds with this nitrogen atom in the solvation shell of the deprotonated model of the dye.
CONCLUSION
The influence of solvation of a molecule of the dye Ind 2 on the absorption wavelength is studied by the quantum chemical calculations using the РСМ method with water solvent. These studies show that Ind 2 is solvated by water molecules primarily by the nitrogen-containing fragment with the participation of the phenolic fragment, while Ind 1 is solvated by the phenolic fragment only. Due to this, these dyes have different mechanisms of interaction with water.
Hydration in position 1 decreases the absorption wavelength (667.13 => 599.12), and protonation decreases it a lot (to 385.7 nm), which corresponds to experimental observations. Hydration in position 2 increases the absorption wavelength (667.13 => 695.49 for a water molecule). The experimentally determined absorption wavelength corresponds to the hydration by a water molecule of position 1 and four molecules of position 2. In general, the phenol fragment (position 1) is hydrated first as it is seen from the fact that a hydrogen atom in the neutral (protonated) form attaches to it. However, hydration of the dinitrophenol fragment (position 2) has a greater influence on the absorption wavelength. In pure water, the dye is present in the form of an anion hydrated by 5 water molecules: NPh−+1H2O(1)+ 4H2O(2), 1 molecule in position 1 and 4 molecules in position 2. The solvation shells of Ind 1 and Ind 2 are studied using the molecular dynamics simulation, which detects specific interactions, namely, hydrogen bonds, between the molecules of the dyes and water. It is also found that Ind 2 attracts water molecules into its solvation shell stronger than Ind 1. The simulation confirms the assumptions made on the basis of the quantum chemical calculations.
-
- Dimrot K, Rneichard C, Sriepmann T, Bohlmann F (1963) Uber pyridinium-n-phenol-betaine und ihre verwendung zur charakterisierung der polaritat von losungsmitteln. Annalen der chemie band.
- Machado VG, Rafaela I, Reichardt C (2014) Pyridinium N‑Phenolate Betaine Dyes. Chem Rev 114(20): 10429-10475.
- Shekhovtsov SV, Omelchenko IV, Dyakonenko VV, Shishkin OV, Allmann R, et al. (2012) Synthesis and crystal structure determination of 2,6-di-tert-butyl-4-(2,4,6-triphenylpyridinium-1-yl)phenolate and its corresponding perchlorate salt. Dyes Pigm 92(3): 1394-1399.
- Schramm ADS, Nicoleti CR, Stock RI, Heying RS, Bortoluzzi AJ, et al. (2017) Anionic optical devices based on 4-(nitrostyryl)phenols for the selective detection of fluoride in acetonitrile and cyanide in water. Sens Actuators B Chem 240: 1036-1048.
- de Melo CEA, Nicoleti CR, Nandi LG (2019) Solvatochromism of new substituted 4-[(E)-(4-nitrophenyl)diazenyl]phenolate dyes. J Mol Liquid 301: 112330.
- Stock RI, Sandri C, Rezende MC, Machado VG (2018) Solvatochromic behavior of substituted 4-(nitrostyryl)phenolate dyes in pure solvents and in binary solvent mixtures composed of water and alcohols. J Mol Liquid 264: 327-336.
- Stock RI, Nandi LG, Nicoleti CR, Schramm ADS, et al. (2015) Synthesis and Solvatochromism of Substituted 4-(Nitrostyryl)phenolate Dyes. J. Org. Chem. 80, 16, 7971–7983.
- Jeong J, Min KS, Kumar RS, Mergu N, Son Y-A (2019) Synthesis of novel betaine dyes for multi chromic sensors. J Mol Struct 1187: 151-163.
- Nandi LG, Facin F, Marini VG, Zimmermann LM, Giusti LA, et al. (2012) Nitro-Substituted 4-[(Phenylmethylene)imino]phenolates: Solvatochromism and their Use as Solvatochromic Switches and as Probes for the Investigation of Preferential Solvation in Solvent Mixtures. J Org Chem 77(23): 10668-10679.
- Serhieieva Y, Zakharov A, Kiyko S (2022) Peculiarities of solvatochromism of 4-[[(2,4-dinitrophenyl)methylene]imino-2,6-diphenyl]phenol and Reichardt’s dye. DFT calculations. Kharkiv Uni Bull Chem Series 38(61): 23-30.
- Etienne T, Michaux C, Monari A, Assfeld X, Perpe`te EA (2013) Theoretical Computation of Betain B30 Solvatochromism using a Polarizable Continuum Model. Dyes Pigm 100(1): 24-31.
- Gaussian 09, Revision B 01, MJ Frisch, GW Trucks et al. Wallingford CT, 2010.
- Калугин ОН, Волобуев МН, Колесник ЯВ MDNAES (1999) программный комплекс для компьютерного моделирования ион-молекулярных систем методом молекулярной динамики // Вест. Харьк. ун-та. Химия. 4 (27): С. 58-79.
- Bartkowiak W, Niewodniczanґski W, Misiaszek T, Zalesґny R (2005) First-order hyperpolarizability of pyridinium N-phenolate betaine dye: Ab initio study. Chem Phys Lett 411: 8-13.
- http://zarbi.chem.yale.edu/ligpargen/index.html
- Jorgensen WL, Tirado-Rives (2005) Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. J Proc Nat Acad Sci 102(19): 6665-6670.
- Dodda LS, Vilseck, JZ, Tirado-Rives J, Jorgensen WLJ (2017) 1.14*CM1A-LBCC: Localized Bond-Charge Corrected CM1A Charges for Condensed-Phase Simulations. Phys Chem B 121(15): 3864-3870.
- Dodda LS, de Vaca CI, Tirado-Rives J, Jorgensen WL (2017) LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res 45(W1): W331-W336.
- Farafonov V S, Lebed AV, Mchedlov-Petrossyan NO (2017) Solvatochromic Reichardt's dye in micelles of sodium cetyl sulfate: MD modeling of location character and hydration. Вісник Харківського національного університету, серія «Хімія» 28(51): 5-11.
- https://upjv.q4md-forcefieldtools.org/REDServer/
- Vanquelef E, Simon S, Marquant G, Garcia E, Klimerak G, et al. (2011) R.E.D. Server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res 39: W511-W517.
- Berendsen HJC, Grigera JR, Straatsma TP (1987) The missing term in effective pair potentials. J Chem Phys 91(24): 6269.
-
 Table 1
Table 1 -
Table 2
-
Table 3


















