Review Article
Role of Apoptosis and Caspases in Fatty Liver Disease “What Did We Miss?”
972
Views & Citations10
Likes & Shares
The pathogenetic mechanisms of liver injury in non-alcoholic fatty liver disease (NAFLD), a condition that is assuming global pandemic proportions and is emerging as the commonest cause of liver cirrhosis worldwide, involves death of liver cells triggering inflammation and causing fibrosis. This apoptosis is driven by inflammasomes, especially - NLRP3, which activate caspase enzymes. Activated caspase-1 releases mature IL-1β and IL-18 in response to various cellular danger signals while Caspase-3 and caspase-7 and other caspases are activated with disease progression, they subsequently cleave a major filamentous protein known as cytokeratin-18 (CK-18)- which is a measurable marker of apoptosis. Caspase inhibition has emerged as a logical strategy to prevent inflammasome activation, and prevent disease progression.
Emricasan, an oral pan-caspase inhibitor, had shown good results in animal models and early phase human studies in halting progressive liver disease. A recent large phase 2b human trial however failed to show benefit in NAFLD.
These recent findings have raised concerns about the role of other caspase-independent forms of cell death, and the need to broaden our understanding and hypotheses from apoptotic to necrotic form of cell death that might best explain the failure of this drug.
Keywords: The pathogenetic mechanisms, NLRP3, Caspase inhibition, Necrotic form
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) comprises a spectrum of disorders that are observable in liver histological slides, ranging from a simple intrahepatic accumulation of fat (steatosis or non-alcoholic fatty liver, NAFL) to various degrees of necrotic inflammation (non-alcoholic steatohepatitis, NASH) [1]. Nearly one third of those with simple steatosis (i.e., NAFL) progress to NASH, while another third with NASH progresses to fibrosis and cirrhosis. Natural history studies show that 2-12 % develop hepatocellular carcinoma [2,3].
Liver injury in NASH involves cell death, inflammation and fibrosis. Lipo-toxicity, resulting from intracellular accumulation of saturated fatty acids, cholesterol, or other toxic lipids; and oxidative injury, which is the result of reactive oxygen species (ROS) production by dysfunctional mitochondria or overactive CYP2E1 are thought to be the major events leading to cell death. Once hepatocytes die, they release danger signals, which activate Kupffer cells and hepatic stellate cells (HSC) resulting in inflammation and fibrosis, respectively [4].
The key cellular pathways that mediate pro-apoptotic signaling initiated by toxic saturated free fatty acids and in steatotic liver are lipid-induced activation of mixed lineage kinase 3 (MLK3) and c-Jun N-terminal kinase (JNK), along with the ER stress response [5]. Saturated fatty acids (such as palmitate) and lyso-phospholipids (such as lysophosphatidyl-choline) can activate death receptors on the hepatocyte cell membrane, resulting in mitochondrial toxicity and activation of caspase-8, which then activates the executioner caspases-3, -6 and -7, leading to apoptosis which is characteristic of NASH [6-8].
Pro-inflammatory cytokine IL-1β has a key role in the development and progression of NAFLD and is involved in all the stages of the disease spectrum- ranging from liver steatosis to NASH and fibrosis. Several mechanisms are able to activate IL-1β in NAFLD, which is produced as an inactive protein and needs proteolytic cleavage to be rendered bioactive. Among these mechanisms of activation, the classical NLRP3 Inflammasome Activation Pathway is most studied for its role in NAFLD and other metabolic disturbance [9].
Inflammasome complexes are typically centered around nucleotide-binding oligomerization domain (NOD)-like receptor proteins (NLRPs). The one with the greatest relevance to NASH is NLRP3. NLRP3 inflammasome is a large, intracellular multiprotein complex expressed in both parenchymal and nonparenchymal cells of the liver. NLRP3 inflammasomes activate caspase-1 and release mature IL-1β and IL-18, in response to various cellular danger signals. Studies have revealed NLRP3 inflammasome activation as an emerging factor contributing to NASH development.
The expressions of NLRP3 components, pro-IL-1β and pro-IL-18, are markedly increased in both mouse models and humans with NASH. Moreover, NLRP3 knockout mice or IL-1α or IL-1β knockout mice are protected from diet-induced liver injury, inflammation, and fibrosis. Another study demonstrates that selective inhibition of caspase-1 alleviates hepatic steatosis, inflammation, and fibrosis in a diet-induced mouse model of NASH. These studies strongly suggest that NLRP3 inflammasome may serve as a potential therapeutic target for the treatment of NASH [8-10].
In Kupffer cells, inflammasome activation and IL-1β secretion can be achieved by treating cells with a saturated fatty acid (SFA) like palmitate, plus a (toll like receptor) TLR-2 or TLR-9 agonist- which is same as exposing them to bacterial endotoxin or products released from fatty acid-treated hepatocytes [10].
Beside this, the IL-1 and TNF secreted by activated Kupffer cells act on hepatocytes by accentuating steatosis and promoting further cytotoxicity. By promoting HSC survival, they also stimulate hepatic fibrosis. In addition, activated Kupffer cells release chemokines that recruit inflammatory macrophages, thus, continuing the inflammatory cycle [11].
The morphological features of dying cells are now well recognizable: they show nuclear and cytoplasmic condensation, blebbing of the plasma membrane, and tearing off of membrane-enclosed particles called apoptotic bodies, taking with them intact organelles and portions of the nucleus. The apoptotic bodies are by engulfed degraded removed by phagocytes, innate immune cells, and HSC. Within the liver, the major cell type to eliminate apoptotic bodies is the Kupffer cell [12,13].
In comparison to the programmed form called apoptosis, necrosis constitutes a pathological or accidental mode of cell death characterized by irreversible swelling of the cytoplasm and distortion of organelles, including mitochondria [12,13].
Liver cell apoptosis can be initiated by different complex pathways, the majority involving activation of the caspases (aspartate‐specific cysteine proteases). Eleven human members have been described. It initiates with the activation of Caspases‐2, ‐8, ‐9, and ‐10, which in turn activate a cascade of downstream effector caspases such as ‐3, ‐6, and‐7. Caspase activation and cleavage of specific cellular proteins or “death” substrates within the cell are the characteristic morphological features that define apoptosis. Apoptosis may therefore be viewed, in biochemical terms, as a caspase-mediated form of cell death. And necrosis is characterized mostly in negative terms by the absence of caspase activation [13]. Caspase-3 and caspase-7 are activated with disease progression and subsequently cleave a major filamentous protein known as cytokeratin-18 (CK-18). Cytokeratin‐18 is an intermediate filament representing about 5% of total protein in the liver, exocrine pancreas, intestine and other epithelial tissues. In vitro experiments have shown that as a consequence of caspase digestion during the intermediate stage of apoptosis, cellular release of cytokeratins 18 fragments into the extracellular space occurs and thus, it can serve as a marker of apoptosis [8, 13-16].
The M30 monoclonal antibody detects executioner caspases which cleave keratin-18 to cCK18 during apoptosis. Also, total cell death can be assessed by the levels offlCK18/M65, which detects both cCK18 and intact keratin-18 [17-19].
It has been seen that if there is sufficient energy (ATP) required for the final execution steps that involve caspase 3/7-mediated cleavage of the cytoskeleton and cell movement, this causes of apoptosis. However, when ATP is depleted, there is no apoptosis, rather necroptosis (caspase 8 is not involved; mixed lineage kinase domain-like pseudokinase [MLKL] is recruited and binds to the receptor-interacting protein 1 [RIP1]/RIP3 complex) or necrosis. And, experiments in rodent livers showing NASH have increased expression of RIP3 (a marker of necrosis) and MLKL (a marker of necroptosis) [19,20].
The pilot study by Wieckowska [20] was the first to measure the levels of caspase‐generated CK18 fragments in patients with NAFLD. Caspase‐cleaved CK18 fragments are significantly higher in NAFLD patients than in controls. In a prospective study examining the utility of apoptosis biomarkers to predict fibrosis in patients with NASH, both full length and caspase-cleaved CK-18 were able to discriminate different stages of fibrosis with healthy controls [19-21].
Serum levels of uncleaved CK-18 are able to distinguish between simple steatosis and NASH, and levels of these molecules correlate with the presence of liver fibrosis [21].
It has been observed that mice genetically deficient in either caspase‐1 (which processes pro- IL-1βto IL-1β), -3, or -8 are resistant to diet‐induced NASH 9. Additional evidence supporting the role of caspase-1 in NASH comes from studies in which NLRP3 inhibition or deletion ameliorated liver inflammation and fibrosis in NASH models [22].
Caspase inhibition has therefore been proposed as a way to prevent inflammasome activation, and thus potentially prevent inflammasome mediated inflammation in numerous liver diseases.
Beneficial effects of various caspase inhibitors have been shown in several models of liver diseases in recent times. Nivocasan (GS 9450) a caspase 1, -8, 9 inhibitor completed phase II study in NASH and IIatrial in chronic HCV, where it was shown to cause reduction in the liver of liver transaminases. However, concerns about adverse effects had halted further studies [23].
Decreases in ALT and cCK18 in various therapeutic studies in NASH patients using pioglitazone and vitamin E were associated with improvements in resolution of NASH, steatosis, lobular inflammation, hepatocyte ballooning, and fibrosis stage [24]. Thus, caspase inhibition may be able to halt or resolve liver damage in NASH as there is reason to believe that excessive caspase‐mediated apoptosis and inflammation are clinically important drivers of progressive liver disease in NASH.
Emricasan (IDN-6556) is an oral pan-caspase inhibitor - which was tested in animal models of acute hepatitis and chronic models of non- alcoholic steatohepatitis (NASH) - and shown to decreased apoptosis, inflammation, fibrosis, cirrhosis, and death. In two NASH models, Emricasan Emricasan reduced inflammation, proinflammatory cytokines, caspase activity, apoptosis, and fibrogenesis. In a murine model of cirrhosis induced by bile duct ligation, Emricasan Emricasan decreased portal pressure and improved survival. Also, it was studied in a rat CCl4 model of cirrhosis and was shown to decrease portal pressure, hepatic vascular resistance, inflammation, and liver fibrosis. These preclinical studies suggested that besides improving hepatocellular function, Emricasan Emricasan may even be effective at decreasing hepatic inflammation and fibrosis and treating portal hypertension [25,26].
Human studies with Emricasan Emricasan in HCV hepatitis with elevated aminotransferases, as well as in patients with liver cirrhosis of different etiologies had showed lowering of ALT levels suggesting anti‐apoptotic and anti‐inflammatory effects [27-29].
In patients with NASH and elevated aminotransferases, Emricasan had previously been shown to decrease caspase-3/7 activity, cCK18, flCK18and serum ALT levels after 4 weeks of treatment. Thus, in NASH patients, pan-caspase inhibition was proclaimed to decrease production of the pro-inflammatory cytokine IL-1β, apoptosis, and decrease liver inflammation and fibrosis [30].
The histologic hallmarks of NASH include hepatocyte steatosis accompanied by ballooning, lobular and portal inflammation, with or without fibrosis. NASH histologic activity is quantified by the non-alcoholic fatty liver disease activity score (NAS) and fibrosis is staged according to the NASH Clinical Research Network (CRN) criteria [31].
In a recent double-blind, placebo-controlled study on 318 patients with NAFLD however, Emricasan treatment with 5 mg or 50 mg for 72 weeks failed to achieve either the primary objective of fibrosis improvement without worsening of NASH or the secondary objective of NASH resolution without worsening of fibrosis or. Further, it was noticed that the pan-caspase inhibitor could have worsened fibrosis and ballooning instead [32]. In another randomized trial of patients with cirrhosis Emricasan was found to significantly reduce mean MELD and Child-Pugh scores in subjects with high Model for End-Stage Liver Disease (MELD) scores (15 or more), compared with placebo, with significant improvement in liver functions.
To assess whether Emricasan lowers Portal Hypertension in patients with compensated cirrhosis, multicenter, open-label study enrolled 23 subjects with compensated cirrhosis and PH (hepatic vein pressure gradient [HVPG] >5 mm Hg). In this study Emricasan was shown to decrease the portal hypertension in those with hepatic vein pressure gradient > 12. However, in a recent a multicenter double-blinded study, Emricasan did not meet either of its primary phase 2b trial endpoints in patients with nonalcoholic steatohepatitis-related decompensated cirrhosis. In this study on 263 patients with NASH-related cirrhosis and baseline HVPG ≥12 mmHg, despite a reduction in biomarkers indicating target engagement, Emricasan was not associated with improvement in HVPG or clinical outcomes.
SUMMARY
In lipotoxic liver injury, hepatocytes die via apoptosis, necrosis and necroptosis. The former is a result of inflammasome activation and is caspase mediated cell death. The theoretically promising aspect of caspase inhibitors was to stop this pathway, as had been shown in earlier animal and human studies.
While suicide-substrate types of caspase inhibitors have been found to be potent and highly effective in preventing apoptotic cell death, the present negative human study in NAFLD raises questions about other factors and mechanisms. It needs to be explored whether inhibition of caspases might increase necroptosis by triggering other caspase-independent forms of cell death [9]. It is therefore possible that caspase inhibitors, though promising in short term studies, could be triggering a more prominent inflammatory response and inducing necrosis when used long-term, and showing up to be not just ineffective but even be detrimental (Figure 1).
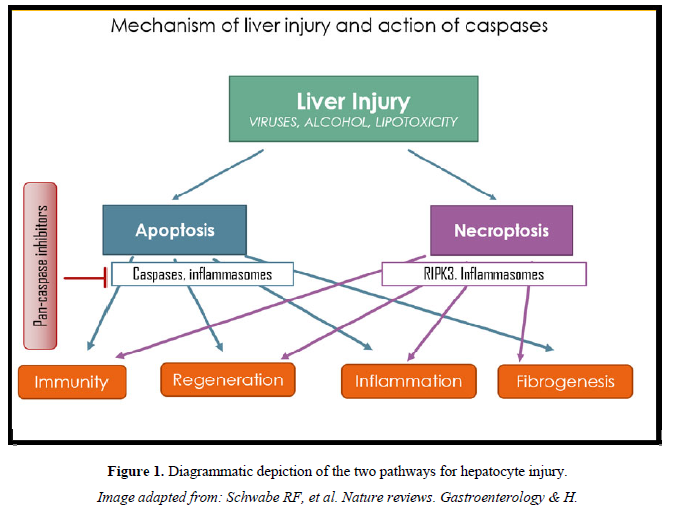
- Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, et al. (2018) The diagnosis and management of non-alcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatol 67: 328‐357.
- Wong VW, Wong GL, Choi PC, Chan AW, Li MK, et al. (2010) Disease progression of non-alcoholic fatty liver disease: A prospective study with paired liver biopsies at 3 years. Gut59: 969-974.
- Yatsuji S, Hashimoto E, Tobari M, Taniai M, Tokushige K, et al. (2009) Clinical features and outcomes of cirrhosis due to non-alcoholic steatohepatitis compared with cirrhosis caused by chronic hepatitis C. J Gasteroentrol Hepatol 24: 248-254.
- Buzzetti E, Pinzani M, Tsochatzis EA (2016) The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD) Metabolism 65: 1038‐1048.
- Hirsova P, Ibrahim SH, Gores GJ, Malhi H (2017) Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. Published correction appears in J Lipid Res 58: 299 J Lipid Res 57: 1758‐1770.
- Marra F, Svegliati-Baroni G (2018) Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol 68: 280‐295.
- Galluzzi L, Lopez-Soto A, Kumar S, Kroemer G (2016) Caspases Connect Cell-Death Signaling to Organismal Homeostasis. Immunity 44: 221-231.
- Akazawa Y, Nakao K (2018) To die or not to die: Death signaling in nonalcoholic fatty liver disease. J Gastroenterol 53: 893-906.
- Mirea AM, Stienstra R, Kanneganti TD, Tack CJ, Chavakis T, et al. (2020) Mice Deficient in the IL-1β Activation Genes Prtn3, Elane, and Casp1 Are Protected Against the Development of Obesity-Induced NAFLD. Inflammation 43(3): 1054-1064.
- Bessone F, Razori MV, Roma MG (2019) Molecular pathways of non-alcoholic fatty liver disease development and progression. Cell Mol Life Sci 76: 99‐128.
- Duwaerts CC, Maher JJ (2014) Mechanisms of Liver Injury in Non-Alcoholic Steatohepatitis. Curr Hepatol Rep 13: 119‐129.
- Schwabe RF, Luedde T (2018) Apoptosis and necroptosis in the liver: A matter of life and death. Nat Rev Gastroenterol Hepatol 15: 738‐752.
- Kanda T, Matsuoka S, Yamazaki M (2018) Apoptosis and non-alcoholic fatty liver diseases. World J Gastroenterol 24: 2661‐2672.
- Woolbright BL, Ding WX, Jaeschke H (2017) Caspase inhibitors for the treatment of liver disease: Friend or foe? Expert Rev Gastroenterol Hepatol 11: 397‐399.
- Yilmaz Y (2009) Systematic review: Caspase-cleaved fragments of cytokeratin 18 - the promises and challenges of a biomarker for chronic liver disease. Aliment Pharmacol Ther 30: 1103‐1109.
- Yang ZH, Yang SX, Qin CZ, Chen YX (2015) Clinical values of elevated serum cytokeratin-18 levels in hepatitis: A meta-analysis. Hepat Mon 15(5): e25328.
- Caulin C, Salvesen GS, Oshima RG (1997) Caspase cleavage of keratin 18 and reorganization of intermediate filaments during epithelial cell apoptosis. J Cell Biol 138: 1379-1394.
- He L, Deng L, Zhang Q, Guo J, Zhou J, et al. (2017) Diagnostic Value of CK-18, FGF-21, and Related Biomarker Panel in Non-alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Biomed Res Int 2017: 9729107.
- Linder S, Havelka AM, Ueno T, Shoshan MC (2004) Determining tumor apoptosis and necrosis in patient serum using cytokeratin 18 as a biomarker. Cancer Lett 214: 1‐9.
- Feldstein AE, Wieckowska A, Lopez AR, Liu YC, Zein NN (2009) Cytokeratin-18 fragment levels as non-invasive biomarkers for non-alcoholic steatohepatitis: A multicenter validation study. Hepatol 50: 1072‐1078.
- Wan X, Xu C, Yu C, Li Y (2016) Role of NLRP3 Inflammasome in the Progression of NAFLD to NASH. Can J Gastroenterol Hepatol 2016: 6489012.
- Vuppalanchi R, Deppe RB, Yates KP (2013) Changes in serum cytokeratin 18 levels significantly predict changes in liver histology in adults with non-alcoholic steatohepatitis: Results from the Pivens trial. Gastroenterol 144: S951.
- Canbay A, Feldstein A, Baskin-Bey E, Bronk SF, Gores GJ, et al. (2004) The caspase inhibitor IDN-6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. J Pharmacol Exp Ther 308: 1191-1196.
- Gracia-Sancho J, Contreras PC, Vila S, Garcia-Caldero H, Spada AP, et al. (2016) The pan-caspase inhibitor Emricasan improves the hepatic microcirculatory dysfunction of CCL4- cirrhotic rats leading to portal hypertension amelioration and cirrhosis regression. Hepatology 64: 1043A.
- Frenette CT, Morelli G, Shiffman ML, Frederick RT, Rubin RA, et al. (2018) Emricasan improves liver function in patients with cirrhosis and high model for end‐stage liver disease scores compared with placebo. Clin Gastroenterol Hepatol pii: S1542-3565.
- Shiffman ML, Pockros P, McHutchison JG, Schief ER, Morris M, et al. (2010) Clinical trial: The efficacy and safety of oral PF‐03491390, a pan caspase inhibitor ‐ A randomized placebo‐controlled study in patients with chronic hepatitis C. Aliment Pharmacol Ther 31: 969‐978.
- Pockros PJ, Schiff ER, Shiffman ML, McHutchison JG, Gish RG, et al. (2007) Oral IDN‐6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatol 46: 324‐329.
- Shiffman M, Freilich B, Vuppalanchi R (2019) Randomised clinical trial: Emricasan versus placebo significantly decreases ALT and caspase 3/7 activation in subjects with non-alcoholic fatty liver disease. Aliment Pharmacol Ther 49: 64‐73.
- Rastogi A, Shasthry SM, Agarwal A, Bihari C, Jain P, et al. (2017) Non-alcoholic fatty liver disease - histological scoring systems: A large cohort single-center, evaluation study. APMIS 125: 962‐973.
- Harrison SA, Goodman Z, Jabbar A, Vemulapalli R, Younes ZH, et al. (2020) A randomized, placebo-controlled trial of Emricasan in patients with NASH and F1-F3 fibrosis. J Hepatol 72: 816‐827.
- Garcia-Tsao G, Fuchs M, Shiffman M (2019) Emricasan (IDN-6556) Lowers Portal Pressure in Patients with Compensated Cirrhosis and Severe Portal Hypertension. Hepatol 69: 717‐728.
- Garcia-Tsao G, Bosch J, Kayali Z, Harrison SA, Abdelmalek MF, et al. (2020) Randomized placebo-controlled trial of Emricasan for non-alcoholic steatohepatitis-related cirrhosis with severe portal hypertension. J Hepatol 72: 885‐895.

QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- International Journal of Medical and Clinical Imaging (ISSN:2573-1084)
- Advance Research on Alzheimers and Parkinsons Disease
- Journal of Neurosurgery Imaging and Techniques (ISSN:2473-1943)
- Journal of Otolaryngology and Neurotology Research(ISSN:2641-6956)
- Journal of Infectious Diseases and Research (ISSN: 2688-6537)
- International Journal of Internal Medicine and Geriatrics (ISSN: 2689-7687)
- Journal of Pathology and Toxicology Research


