3697
Views & Citations2697
Likes & Shares
Small molecule RNAs (small RNAs, sRNAs) are regulatory factors consisting of 20 to 24 nucleotides (n) that regulate gene expression at the transcription and post-transcribed levels. sRNAs mainly realize the purpose of regulating biological processes by regulating the translation process of mRNA transcribed. sRNAs play a key regulatory role in plant growth, development, metabolism, and plant anti-reversal reactions. Depending on the composition and functionality of sRNAs, it can be divided into siRNAs (small indin RNAs, siRNAs) and miRNAs (microRNAs) which are more conservative and tissue specific.
INTRODUCTION
Plants have constantly been exposed to numerous abiotic and biotic constraints. One important abiotic stress is metal toxicity. Both metals that are essential for plant growth and development such as copper, iron, zinc and manganese, and non-essential counterparts; cadmium, aluminum, cobalt, mercury to mention but a few can be hazardous as high concentrations of any metal type is toxic for the plant. Mercury pollution has become increasingly serious in recent years, from the beginning of human industrialization to 2000, resulting in 640 x 106 kg of mercury emissions [1]. Mercury is present in soil in many forms, where ion mercury (Hg2 plus) can be absorbed by plants due to its soluble and movable properties [2]. Although plants absorb less mercury, they are more toxic due to mercury [3], a small amount of absorption can also cause significant damage. Mercury damage to plants is mainly manifested in two aspects: 1, directly binding to biological macromolecules such as protein-based, thereby disrupting the function of biological macromolecules, disrupting the normal structure of the mass membrane. 2. Heavy metal damage generally stimulates an increase in reactive oxygen (reactive oxygen species, ROS) levels in plants, leading to oxidative stress, which is a major cause of plant damage [4]. In addition, mercury inhibits seed germination, causing damage to photosynthesis organs, reducing yield and even plant death [5].
In response, plants have deployed various protective strategies that include the synthesis of different proteins involved in detoxification, such as phytochelatins and metallothioneins. Exudation of organic acids that easily form complexes with heavy metals via the root system have been some key physiological strategies [6].
The metal transporters expression has been deemed core to heavy metal toxicity tolerance [7]. Small and/or large non-protein coding RNAs (npcRNAs) have been hypothesized to be involved in regulation/signaling of metal toxicity response. One of the most studied classes of npcRNAs is the micro RNAs (miRNAs). miRNAs are 21 nucleotides in length npcRNAs that regulate post-transcriptional level gene expression in plants. A precursor miRNA (pre-miRNA) with imperfect hairpin structure is processed into a mature miRNA and this is incorporated into an RNA-induced silencing complex (RISC) that promotes degradation/cleavage of the corresponding target mRNA(s), recognized by an almost perfect base complementarity with the miRNA. Though most of the miRNAs identified in Arabidopsis thaliana are related to plant development, there is evidence of the role of miRNAs in the plant respond to different abiotic stresses including metal toxicity [8].
Metal toxicity stress triggers a surge in the antioxidative enzymes and their activities resulting into unbalancing reactions of these important proteins due to high accumulation of ROS (reactive oxygen species). This causes oxidative stress leading to damage of various biomolecules such as lipids, proteins, and DNA [9]. Understanding the ability of plants to receive stress signal and create an appropriate response is key in plant breeding [6].
MATERIALS AND METHODS
Download, collect and organize Tribulus data resources
sRNA Library
Download 46 Tribulus sRNA libraries [10-16], these libraries cover different tissues of Tribulus quinoa, including seedlings, root, root tumor, root cancer, leaves, flowers, seeds, etc., as well as under different conditions including rdr6 mutation, plexus root symbiosis, drought stress, Hg stress, etc., more comprehensively contain aquinopha sRNAs. RDR6 is one of the essential factors for producing endogenous siRNA, and rdr6 mutation libraries will help us distinguish between miRNA and siRNA. Using the MATLAB software design program to organize and merge the sRNA sequences in these libraries.
Degradation Group (degradome) Library
Similar to sRNA, download 3 Tribulus degradation libraries from the NBCI GEO website [12,14], combined with the laboratory has been sequenced in the early 2, a total of 5, these libraries related to the quinoa root, leaves, flowers and other tissues and Hg stress and plexus root symbiosis and other growth conditions, their comprehensive use, can be more effective in the elimination of false positive targets, improve the accuracy of identification.
Genomics and Gene Pool
Download the Tribulus genome chromosomal sequence and note gene sequence (Mt4.0v1: http://medicago.jcvi.org/medicago/index.php) containing 8 chromosomes, 1,141 scaffolds, 62,319 There are 1,170 genes that are marked as disease-resistant proteins, including 550 in the TIR-NBS-LRR category, 124 in the CC-NBS-LRR category, 396 in the NB-ARC category, and 100 of other types.
miRNA
Download 756 miRNAs from the miRBase website (http://www.mirbase.org/index.shtml;miRBase 20).
New miRNA Identification
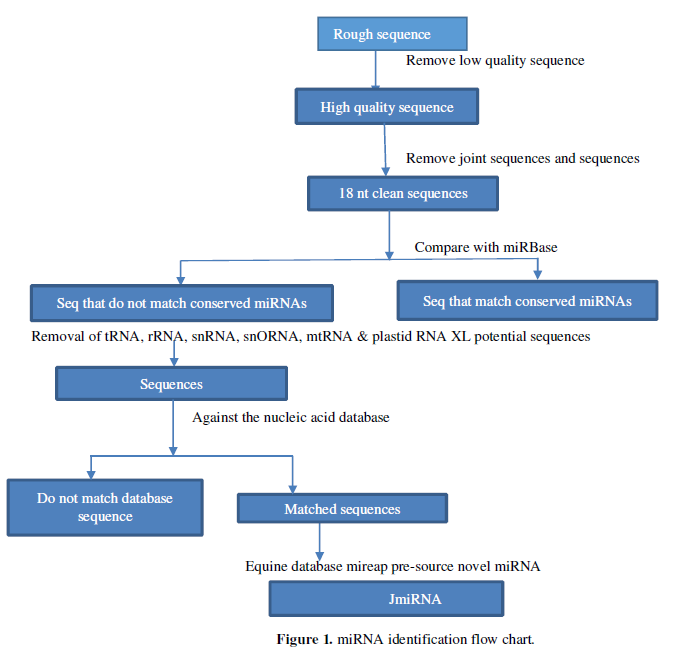
Based on previous literature on high-throughput sequencing for miRNA analysis, the specific process of identifying miRNA in this experiment is shown in Figure 1:
- Integrate the downloaded sequencing database, at which point the sequence contains the sequencing joint, which contains a lower mass sequencing base, so the first thing to remove the connector sequence in the raw data, low-quality sequence, and the miRNA is generally longer than 18nt, so the sequence shorter than 18nt is also filtered out, leaving the so-called “Clean sequence” go to the next analysis
- “Clean Sequence” includes rRNA, known miRNA, etc., so first remove the known miRNA by compared the known miRNA in miRBase to the “clean sequence” of the previous processing, setting up with the known miRNA Only two sequences with the following mismatches are considered conservative miRNA
- In addition to the known miRNA, there are snRNA, tRNA, etc. in sRNA, so the sequence that cannot be matched with the miRBase database is compared to the snRNA, rRNA, tRNA, and plasmatic RNA database and mitochondrial RNA database, that is, the classification notes the sequence measured sRNA, the remaining unnoteable sequence, that is, considered to be the possible sequence of the new miRNA, into the next analysis process
- In order to avoid false positives, the new possible miRNA sequence will also be compared with the Tribulus Genome Database (M. truncatula Genome release 4.0) to extract sequences matched with small RNA in the Tribulus nucleic acid database. Predict its secondary structure and the characteristics of Dicer enzyme cut points, folding free energy. This step is implemented by mi reap software, through the screening criteria corresponding to the small RNA sequence into the next round of screening.
- In order to distinguish miRNA and other endogenous sRNAs, the transcription sequence of miRNA can produce a special secondary structure of the properties, intercept the genome sequence of the upper and lower reaches of the sRNA has been compared, and obtain the secondary structure details of miRNA using mfoldRNA software. Minimum free energy, secondary structure and other information, to examine its secondary structure information, mainly based on: (1) in the prediction of folding miRNA precursors can be found in miRNA and the corresponding miRNA. (2) MiRNA and the opposite miRNA are located on the arm of the predicted forebody, not on the ring, and at the 3 end there should be 2 bases protruding. (3) Not every miRNA and miRNA pair are perfect, but the number of mismatches should not exceed 4 bases. (4) On the arm of the hairpin structure, the protrusion of 2 bases cannot be greater than 2 bases when there is a protruding structure on one side. The small RNA sequence after screening is considered to be the new miRNA. Each of these data processing stakes in quality control and screening standards to ensure that the data goes to the next step is reliable.
Combined with degradation group data analysis to predict miRNA targets
The determination of plant miRNA targets is the basis of its functional research, and plant predictions are more reliable than in animals due to the high degree of complementarity between plant miRNA and its target mRNA. The prediction rules for miRNA and targets commonly found are as follows: (1) miRNA and target up to 3 mismatched bases; 2-12 bit base and target up to 2 mismatches; (4) the minimum free energy of miRNA and target dipolymer: miRNA and target binding bit dipolymer minimum free energy ratio should be greater than 0.75. Based on four principles similar to the above, some plant miRNA target prediction software has been developed.
Here, three online software predictions are used: WMD3 (http://wmd3.weigelworld.org/cgi-bin/webapp.cgi), psRNATarget (http://plantgrn.noble.org/psRNATarget/) and UEA plant sRNAtoolkit (http://srna-tools.cmp.uea.ac.uk/plant/cgi-bin/srna-tools.cgi). The usage methods and parameters of the 3 predictors are set as follows:
- PsRNATarget use, after entering the home page, find or paste the text input box below it, paste the miRNA sequence in, find Select a preloaded transcript/library for target search: in the drop-down option below it, select the transcription group 4.0V1 version of the slug, select the default, and then click on the submit sequence, other default parameters, submit the target of the project, and submit the target in a few minutes. The drawback of this method is that the predicted target is cut in the coding area. The others were missed.
- Use of WMD3, go to the home page, find Target Search, click Target Search, you can see a square text input box, will predict the target miRNA sequence paste in, transcript selection. The selection location of the transcript is located in the list box to the right of Genome, where you select the transcription group version 3.5 of the transcription, default to other parameters, submit the query sequence, and generate the predicted structure in a few minutes.
- The use of UEA plant sRNA toolkit, after entering the home page of the UEA plant sRNA toolkit, click on The Target prediction, paste the miRNA sequence to be predicted, Select the Tribulus quinoa transcription group in the drop-down list box below the select transcription group, and the other parameters default, submit the search, and the miRNA target can be obtained in a few minutes.
- Integrate the miRNA targets predicted by the above procedures, combined with the comprehensive analysis of the degradation group library, where the target ratio of miRNA prediction is compared to the genome of Tribulus, which is due to the results of the degradation group data being compared to the genome, in order to facilitate the integration of the two data together, Regardless of the prediction of biological information or the sequencing results of the degradation group data, there will be a predicted cutting site, if the two sites are the same, that is, the program predicts the miRNA cutting site and the cutting site analyzed by the degradation group is consistent, then such miRNA sequence and target is selected, that is, the potential target cutting point is found. This analytical method is a large-scale target identification technique combining high-throughput technology with bio-information means, which not only gets rid of the problem of false positive high brought about by bio-information prediction, overcomes the problem of time-consuming of low throughput of conventional experimental identification targets, and significantly improves the functional research of miRNA.
RESULTS AND ANALYSIS
New miRNA Identification Results
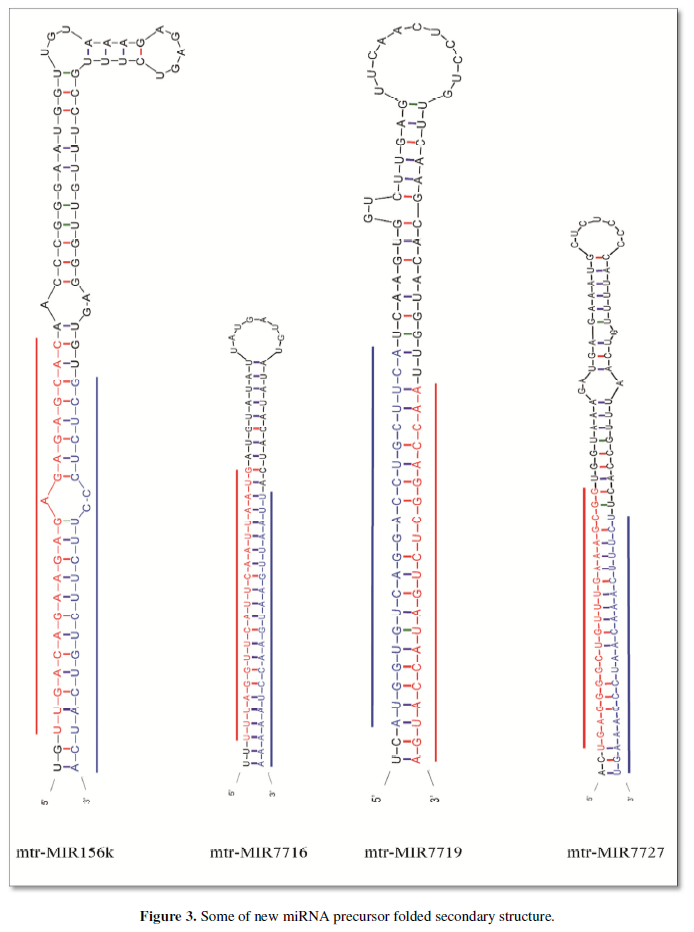
After the small molecular RNA classification annotation is complete, the possible new miRNA is compared to the genome of the quinoa (mi reap software), intercepting sequences that can be folded into stem ring structure, where sRNA and genome sequences are selected for sequences with less than 4 base mismatches, and the smallest folding free energy of the stem ring is formed. Less than -25kcal/mol, miRNA is an important evidence to prove that sRNA belongs to miRNA, in the analysis of the general first to miRNA and miRNA in the distribution of pre-miRNA to eliminate the interference of the variant, generally miRNA and miRNA is generally concentrated at both ends of pre-miRNA, Figure 2 lists some of the distributions of eligible sRNAs on their pre-miRNA. It can be seen that the distribution of these sRNA pre-RNA is concentrated at both ends, and the values to the right of the sequence are corresponding to the sequenced sRNA readings. The precursor sequence left behind in the previous step is folded by the Mfold software, and if the miRNA identification criteria are met: (1) miRNA and the corresponding miRNA can be found on the predicted folded miRNA precursor. (2) MiRNA and the opposite miRNA are located on the arm of the predicted forebody, not on the ring, and at the 3 end there should be 2 bases protruding. (3) Not every miRNA and miRNA pair are perfect, but the number of mismatches should not exceed 4 bases. (4) On the arm of the hairpin structure, when there is a protruding structure on one side, it cannot be greater than the protrusion of the 2 bases is considered to be possible miRNA, Figure 3 is the result of several secondary structural folding images that may be the precursors of miRNA, and marked red with miRNA opposite the miRNA is misplaced to the miRNA of two bases. So far sRNAs that meet the above criteria are considered to be new miRNA. All miRNA precursors that meet the above criteria can be found in Appendix II.
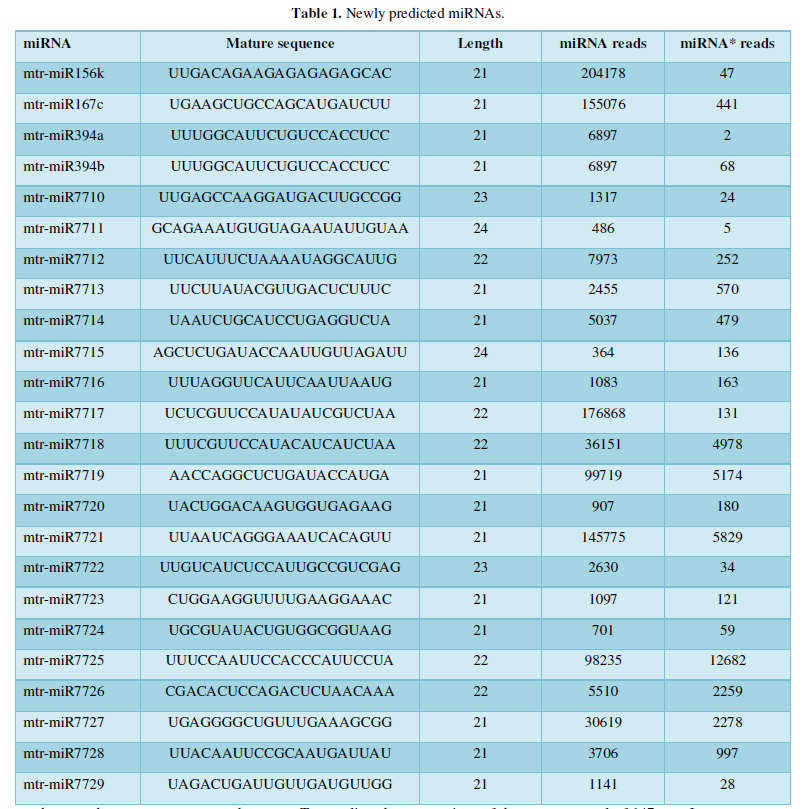
After a few steps of screening, a total of 24 new miRNAs of Tribulus quinoa were identified here, and Table 1 is the ID and sequence of the new miRNA identified according to the identification criteria of plant miRNA. The next step is to predict the target of the new miRNA based on these sequences.
miRNA target prediction results
First of all, the 3 in-line plant target prediction tool prediction target series, and then combined with the degradation group data to analyze miRNA target, it is worth noting that bioinformatics prediction targets have many false positives, so to combine the results of degradation group sequencing to analyze, the 3 program prediction miRNA targets in the degradation group library search, because the data of the prediction of the mi-target RNA is compared to the miRNA to the mi-s Search the quinoa sequence database to obtain the target corresponding genome sequence,

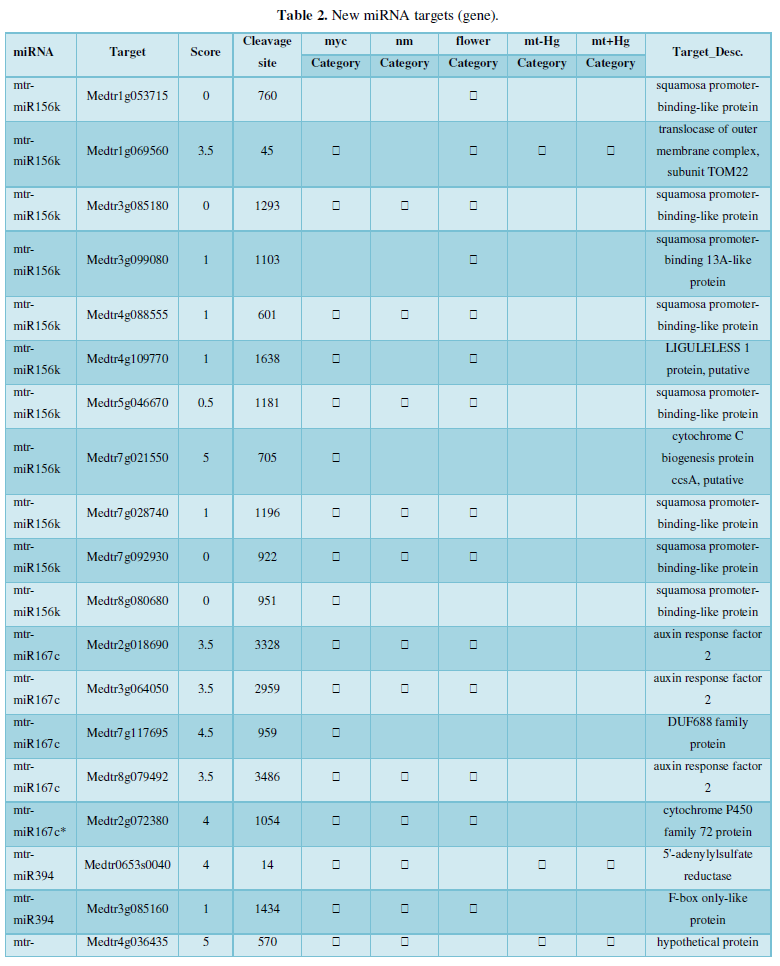
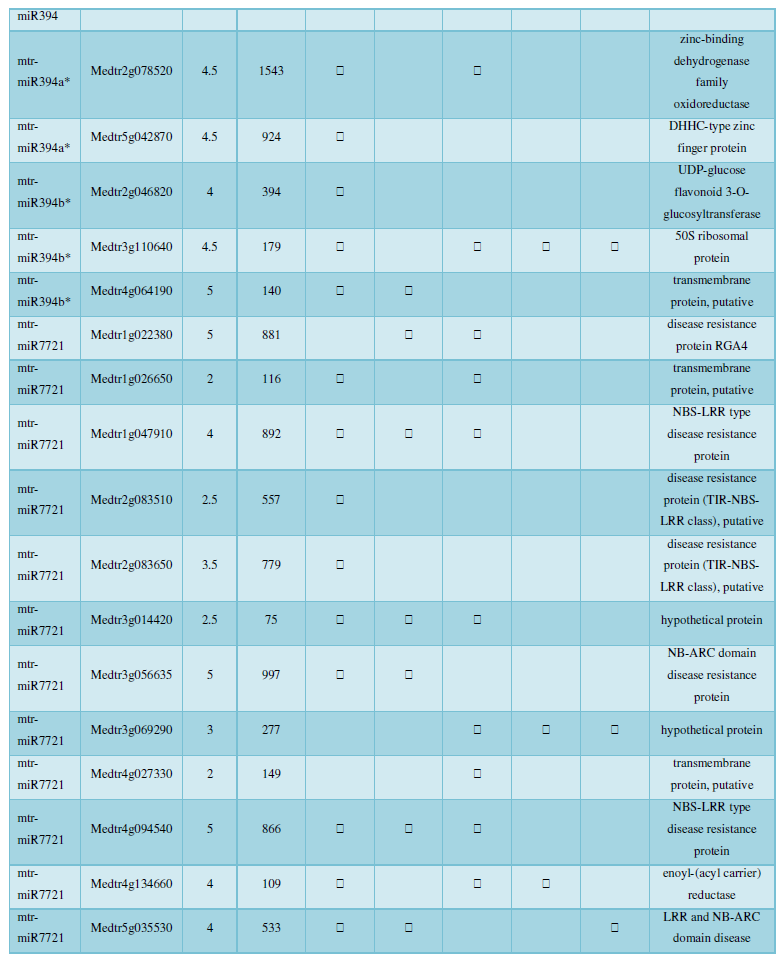
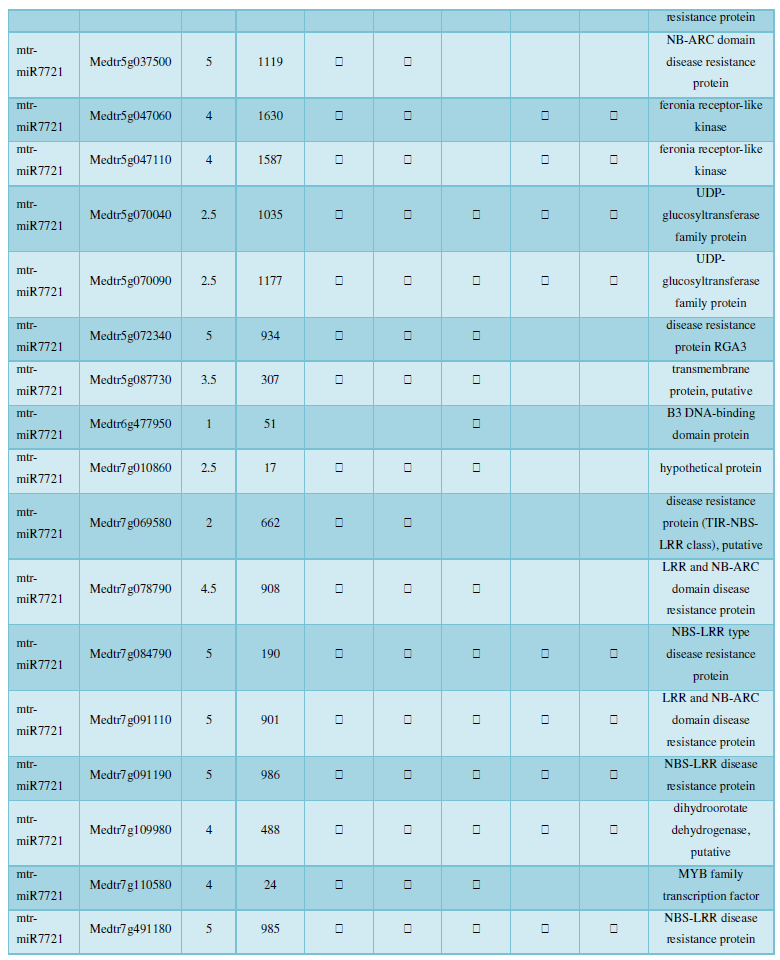
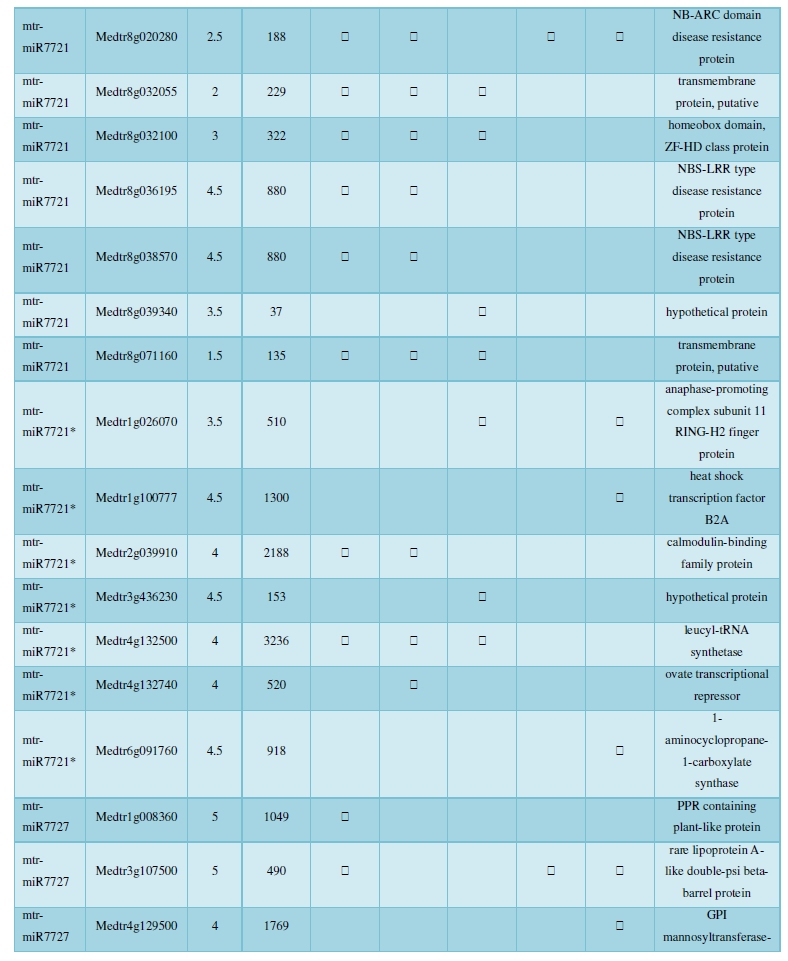
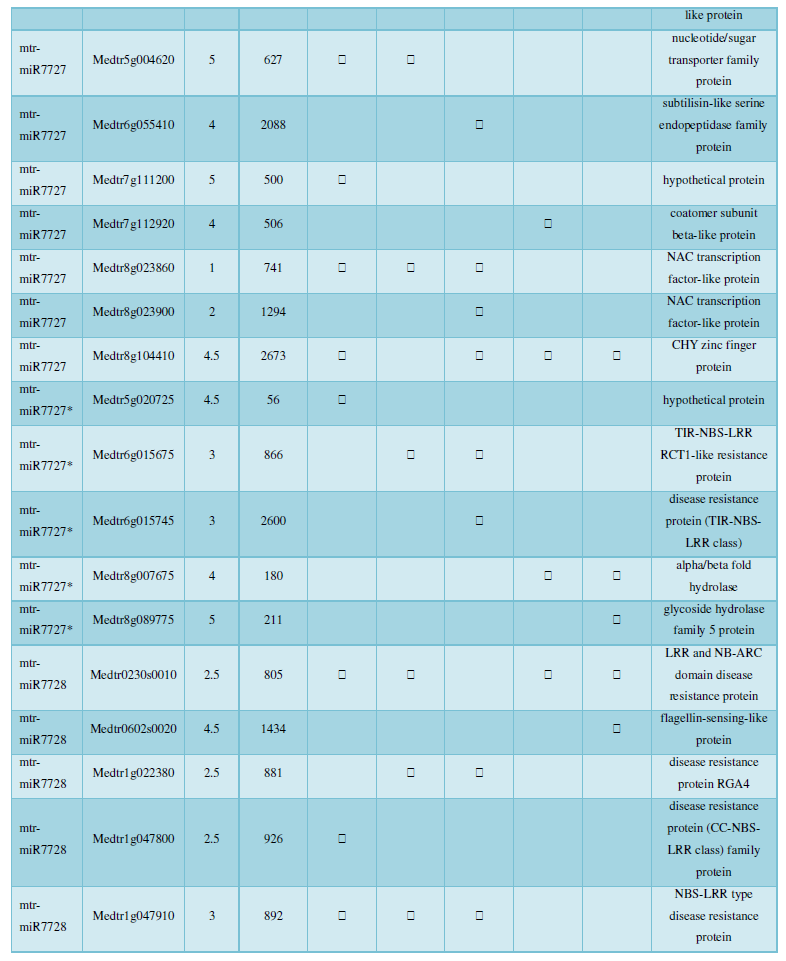
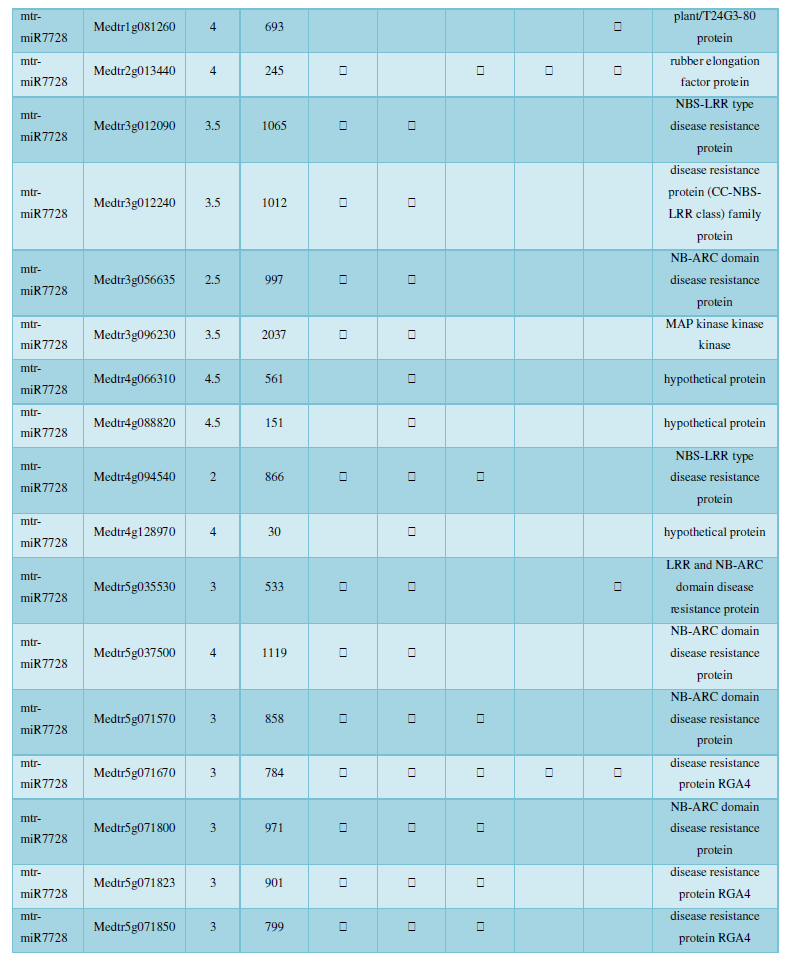
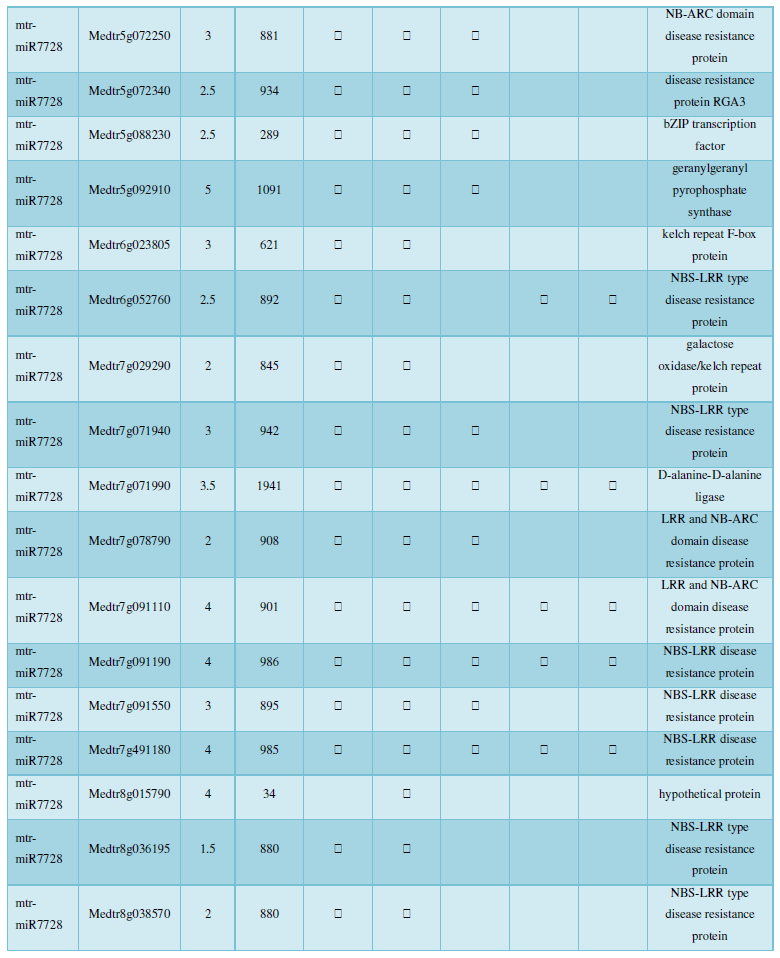
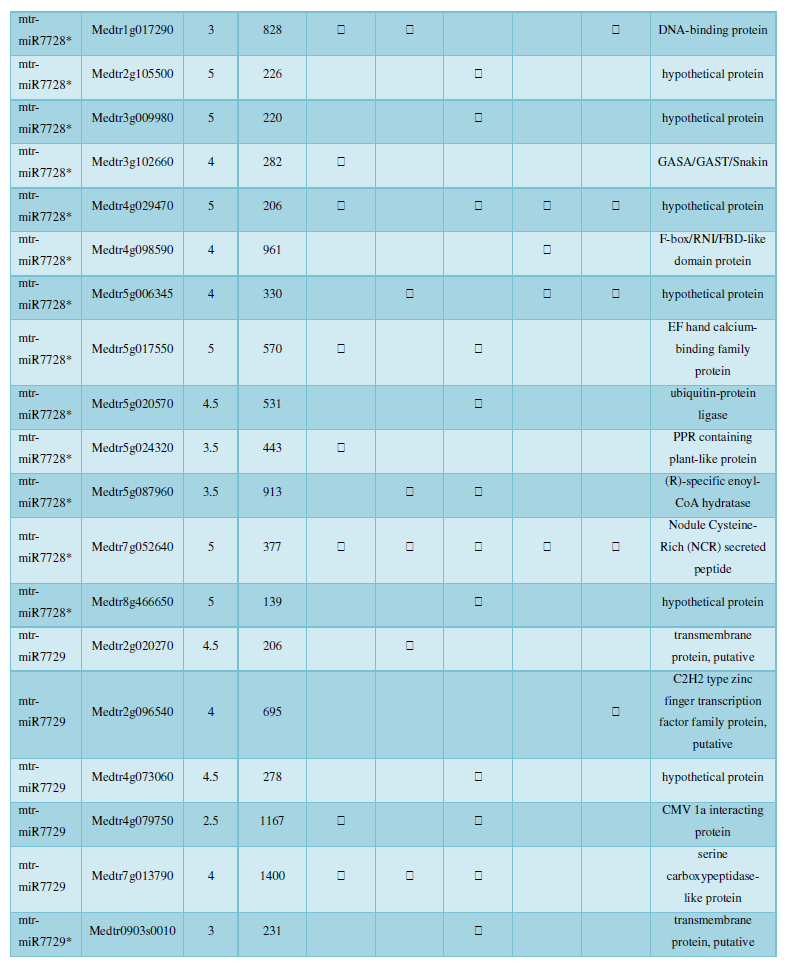
and then use the Target-aligned (http://www.leonxie.com/targetAlign.php?type?blast) online program to predict the matching position of miRNA on the target corresponding genome sequence, and select the predicted miRNA shearing site with the target of the mRNA degradation point in the degradation group, as the target of the miRNA. Table 2 is a combination of miRNA targets predicted by the degradation group data and some annotations of the targets, a total of 147 type I target genes found in miRNA and miRNA.
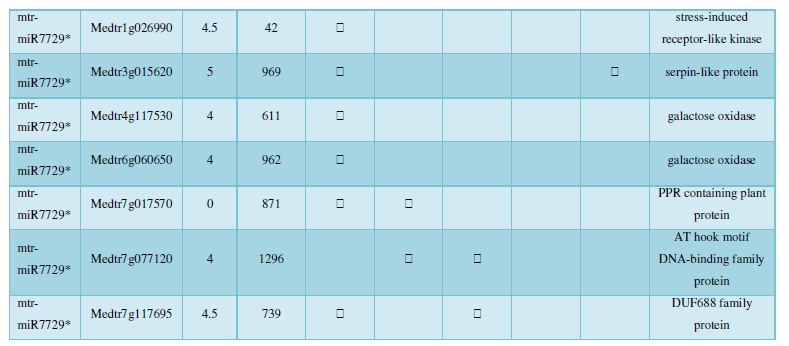
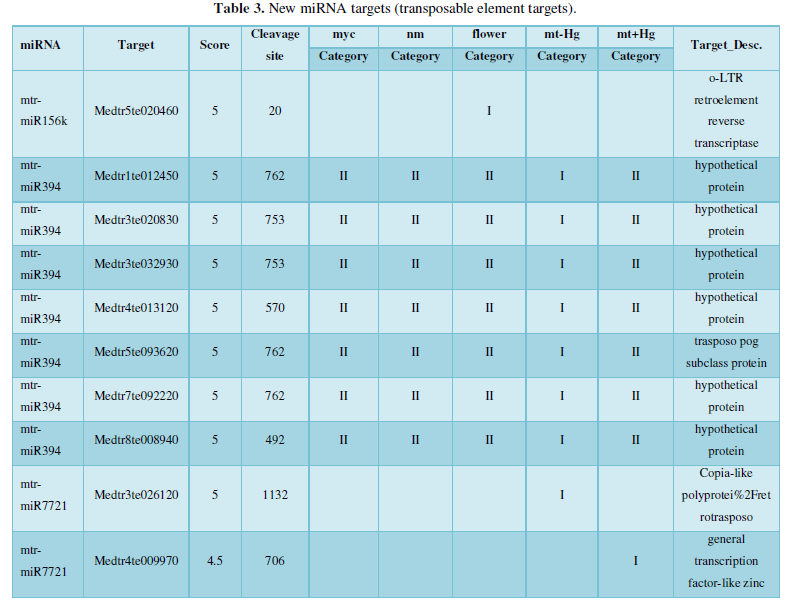
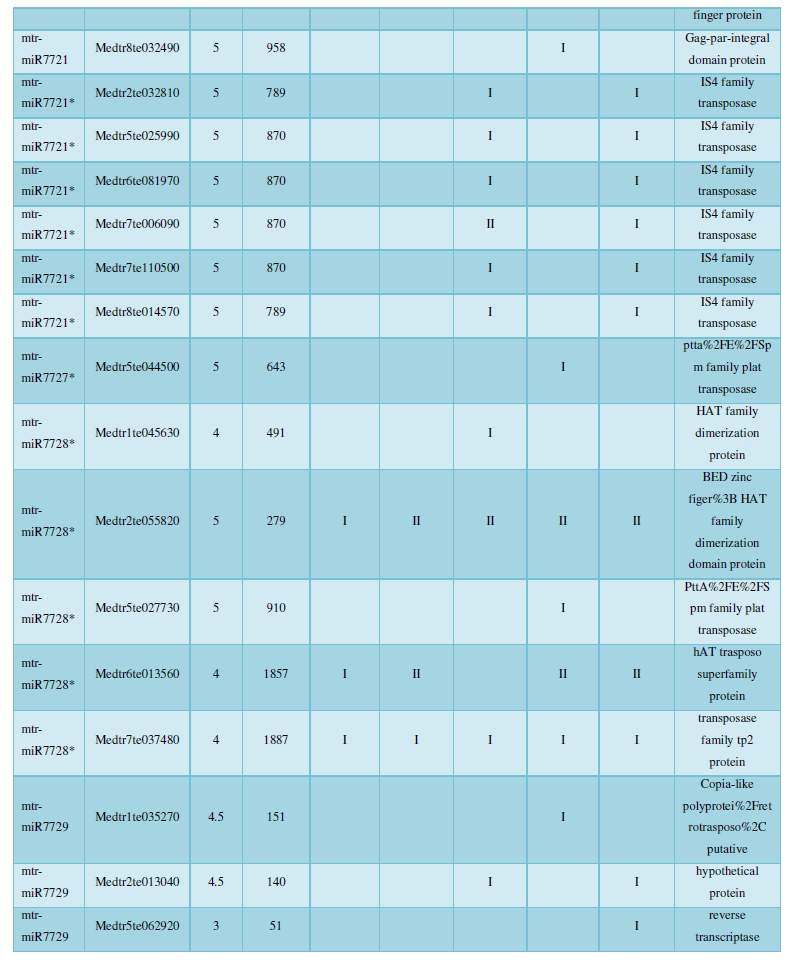
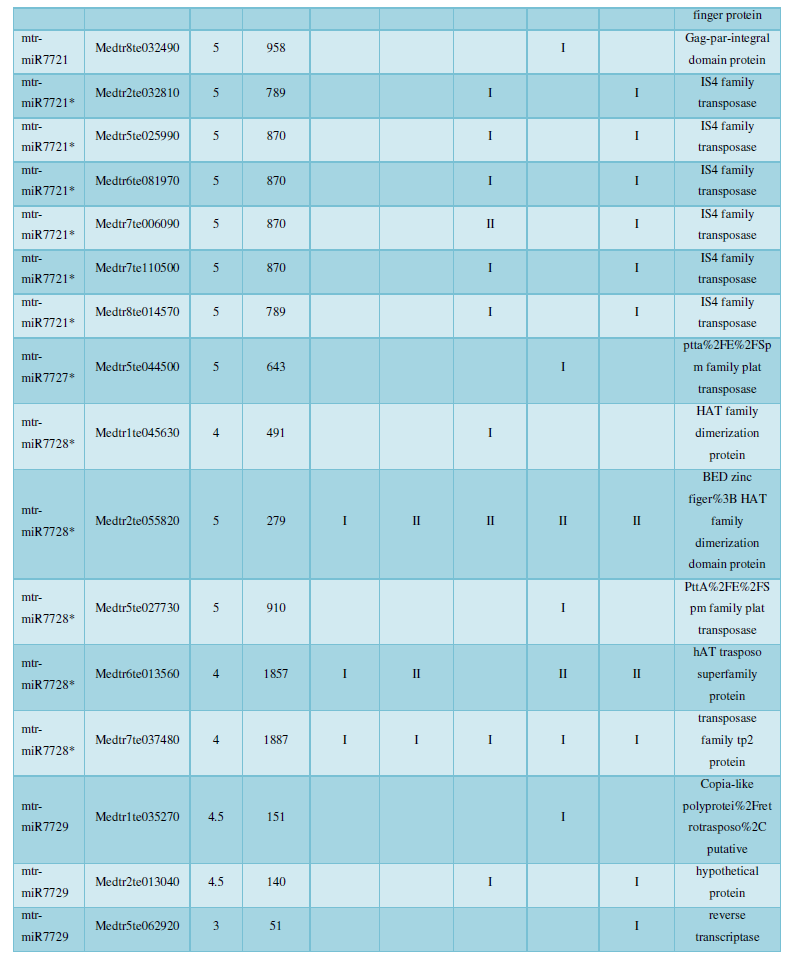
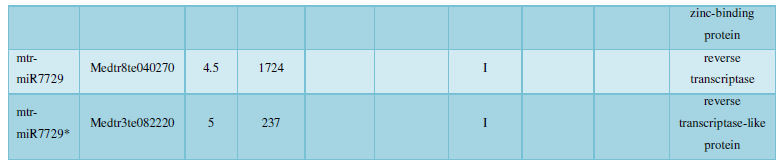
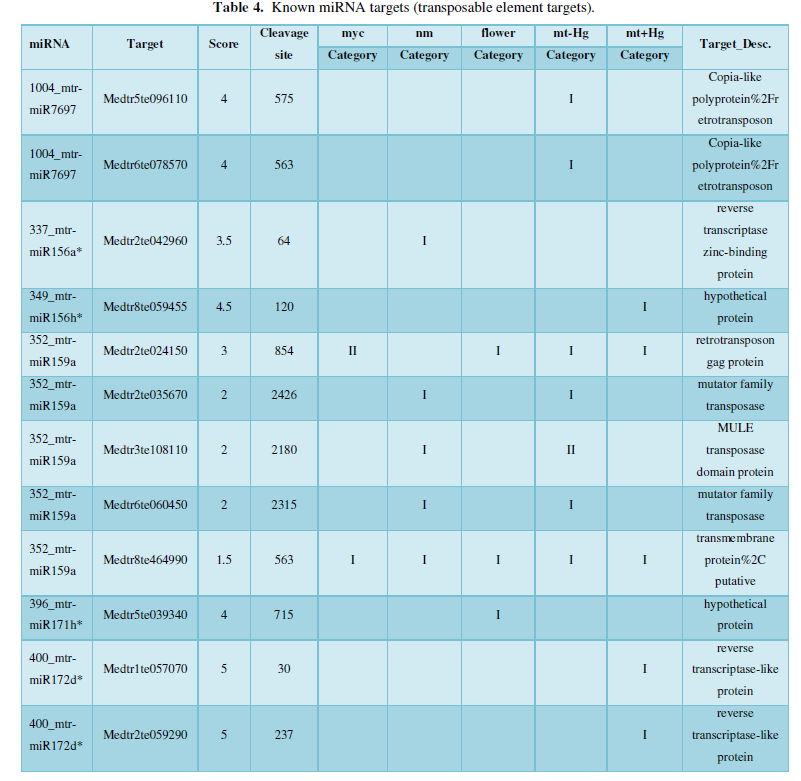
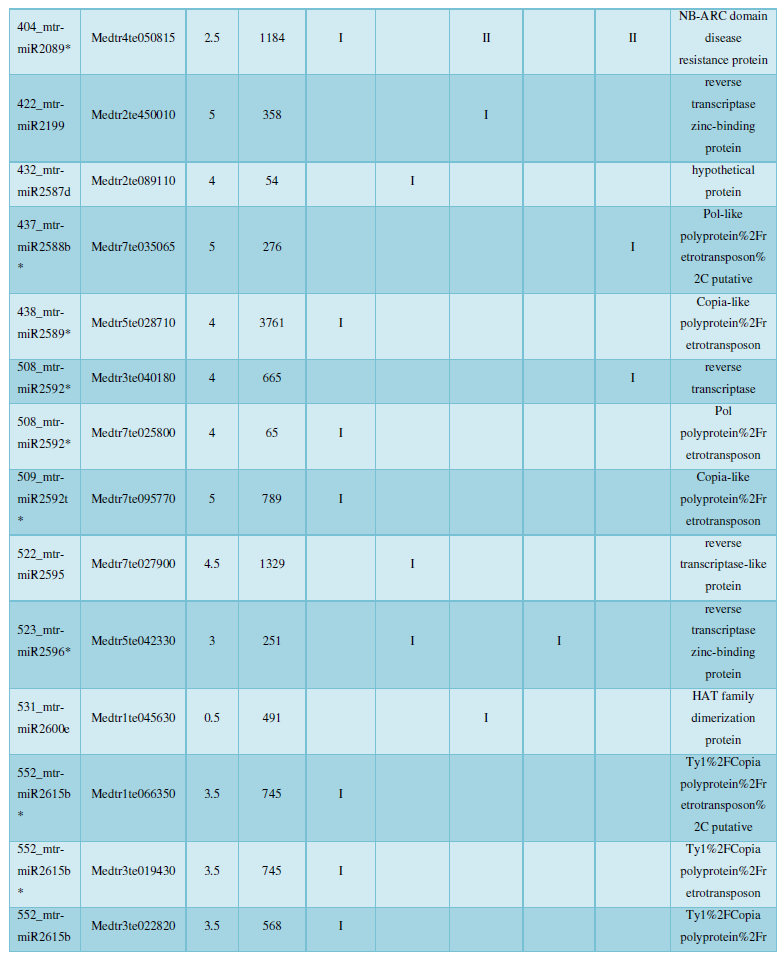
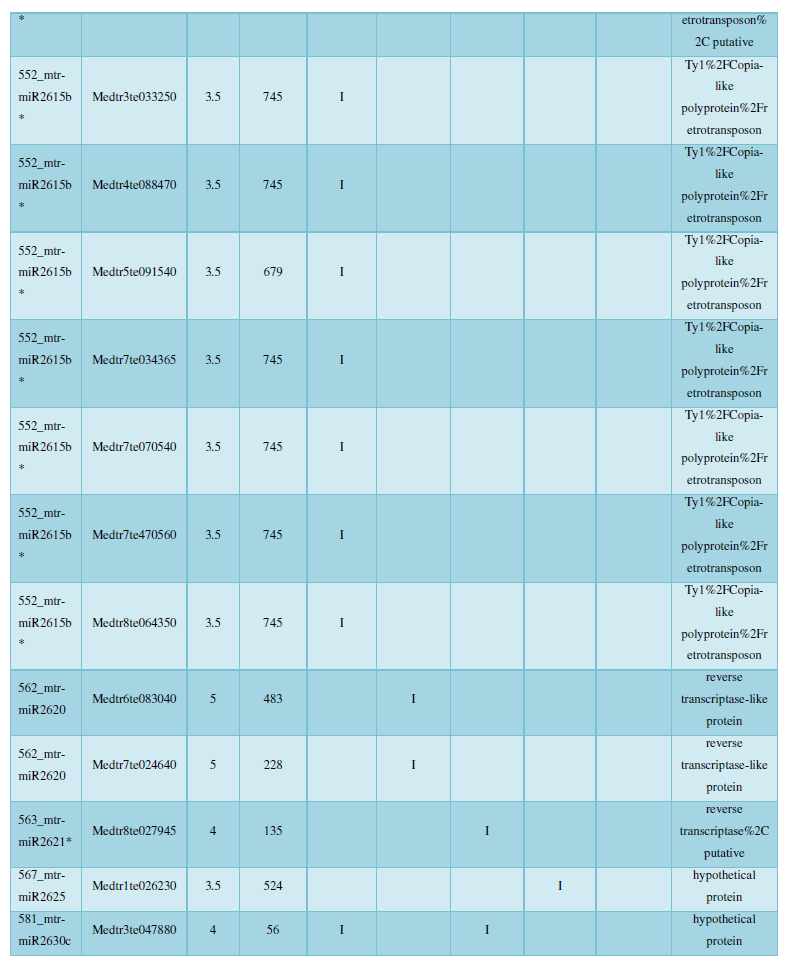
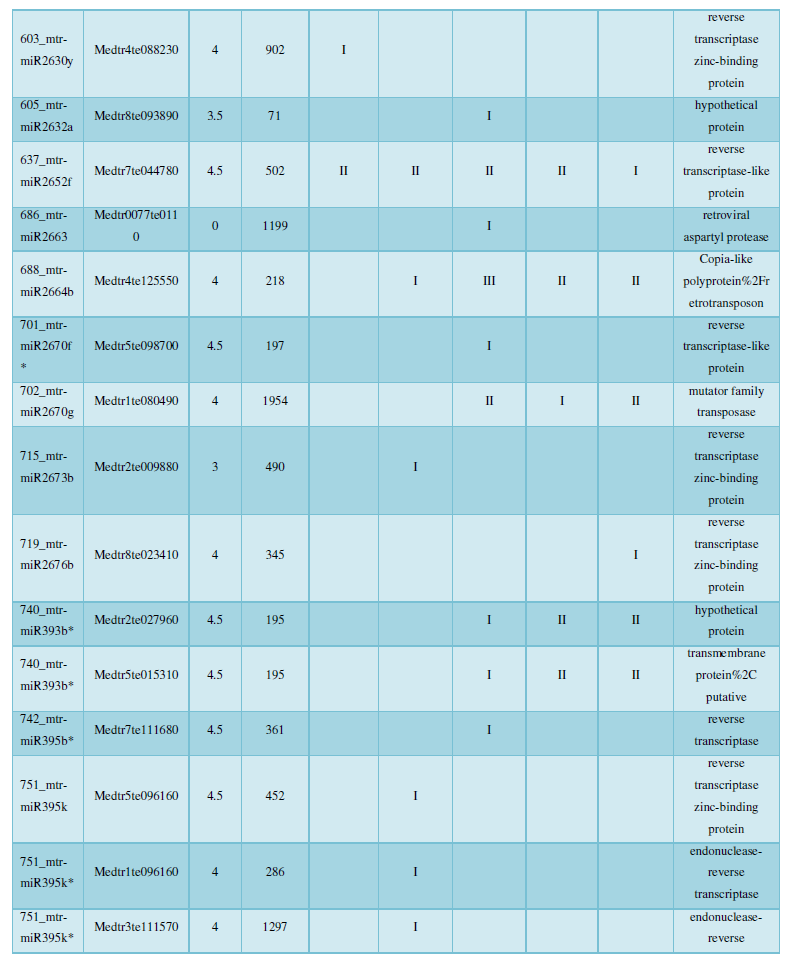
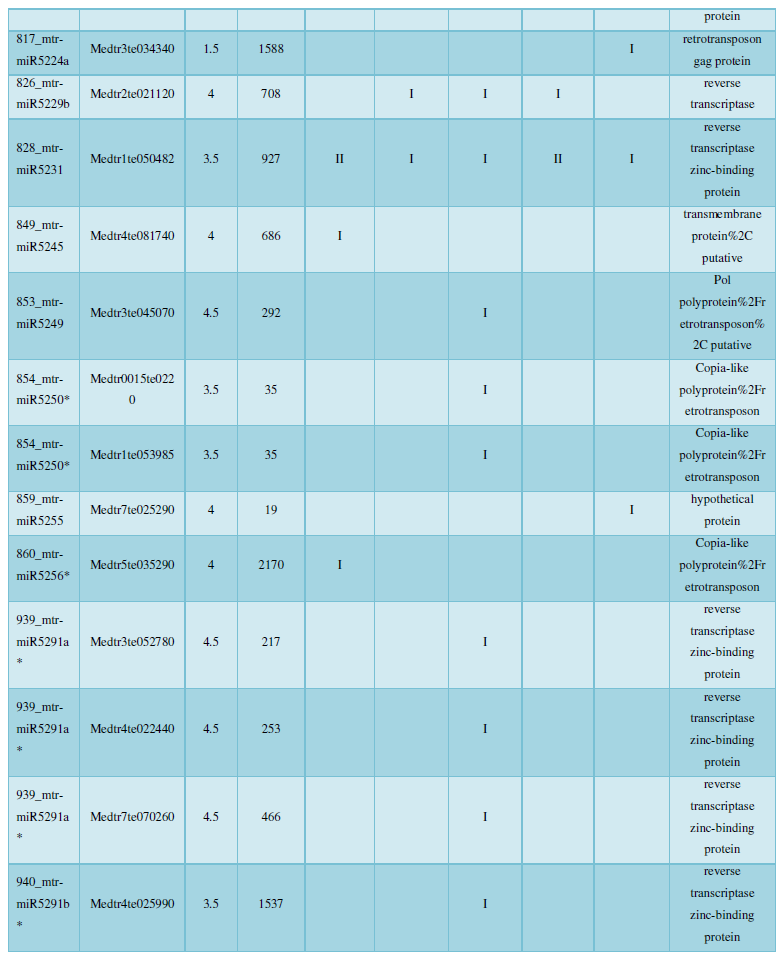
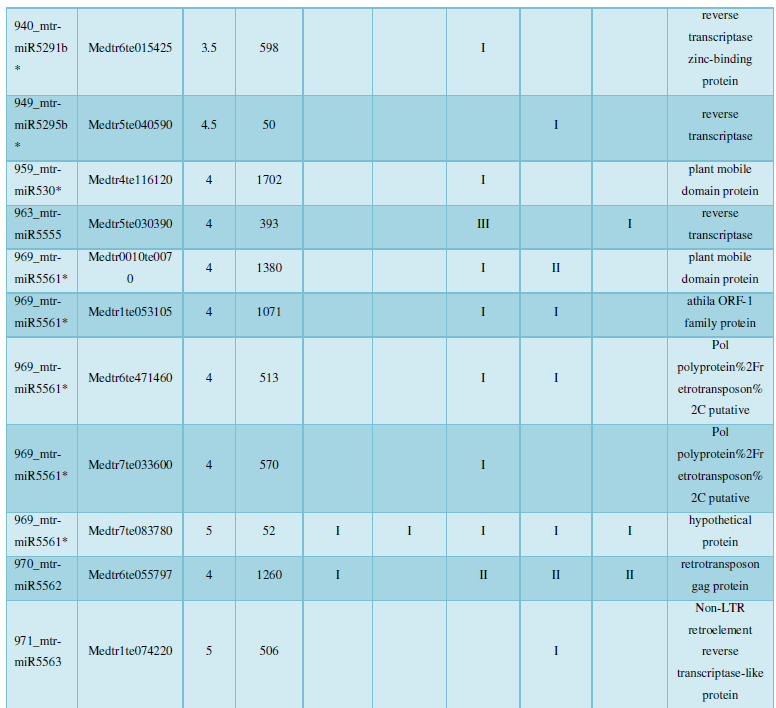
A similar method was used to predict the newly discovered miRNA, miRNA, and the already miRNA, miRNA, and the transposon targets in Tribulus, Table 3, which are the targets of newly discovered miRNA and miRNA, which have a total of 28 type I transposon targets. Table 4 is a rotor target for tribulus quinoa that already has miRNA and miRNA, for a total of 91.