
4009
Views & Citations3009
Likes & Shares
Introduction: The renal complications of sickle cell disease have a spontaneous evolution towards chronic renal failure, with the need for dialysis or renal transplantation. Renal damage is frequent, early and disabling; it is a major cause of mortality. It appears as early as the first decade, it is most often asymptomatic, evolves insidiously and is sometimes discovered by chance. The aim of our study is to describe the urinary profile of patients living with sickle cell disease and in acute crisis.
Patients and method: This are a descriptive cross-sectional study, conducted over a period of four months (from 01 August to 29 November 2019) at the National Reference Centre for Sickle Cell Disease. It focused on homozygous sickle cell patients admitted for an acute crisis. Urine dipstick was performed in each patient, and we assessed parameters such as pH, leukocyturia, hematuria, proteinuria, ketonuria and nitrites.
Results: 219 patients were included in the study (58 % female and 42 % male). The mean age was 20 ± 11 years with extremes of 3 and 59 years. Acute hemolysis crisis was the most frequently encountered emergency (49.3 %). 27.9 % of patients had positive proteinuria; 21.9 % had hematuria; 11.9 % had leukocyturia; 3.2 % had positive nitrites on urine dipstick.
Conclusion: The urine dipstick is a simple means of screening for renal damage in sickle cell disease. It should be performed routinely in all patients seen in acute crisis or during routine visits.
Keywords: Urinary profile, Sickle cell disease, Acute crisis, Brazzaville
INTRODUCTION
Sickle cell disease is a single mutation genetic disease with an autosomal recessive transmission related to a qualitative abnormality in the structure of hemoglobin, which results in the formation of hemoglobin (Hb) S [1].
Under favorable conditions such as hypoxia, dehydration, acidosis or infection, this abnormal hemoglobin polymerizes in the red blood cell [2], causing rheological changes, rigidification and falciformation; origin of acute and chronic complications [3], with repercussions on the organs, including the kidneys. The renal manifestations have a spontaneous evolution towards the installation of a chronic renal insufficiency, with the necessity of dialysis or even renal transplantation [4,5].
Renal damage in patients with sickle cell disease is frequent, early and disabling; it is a major cause of mortality. It appears as early as the first decade [6-10]; its global prevalence is estimated at 25% in children aged 2 to 8 years [7-10].
In young adults with sickle cell disease, the prevalence of renal damage reaches 40% with in half of the cases a rapid progression to end-stage renal failure [7,11-13].
Renal damage is most often asymptomatic, evolving insidiously and sometimes being discovered sometimes discovered by chance.
The aim of our study is to describe the urinary profile of patients with homozygous sickle cell disease admitted in acute crisis.
PATIENTS AND METHOD
This is a descriptive cross-sectional study conducted over a period of four months (from August 1 to November 29, 2019) at the National Reference Center for Sickle Cell Disease (NRCSCD) “Antoinette SASSOU NGUESSO” in Brazzaville. It focused on sickle cell patients admitted to the emergency room for an acute crisis (vaso-occlusive crisis, anemic crisis, fever, etc.). All homozygous sickle cell patients were included in the study. Any patient who came for a routine check-up (who was in apparent good health) was excluded. We systematically performed a urine dipstick (UD) on freshly passed urine collected in a clean jar, and assessed pH, leukocyturia, hematuria, proteinuria, ketonuria, and nitrites in each patient. Data collection was done using a survey form, the patient follow-up form, and the NRCSCD emergency room consultation log. Data entry was done using Excel 2013 software. R software version 4.1.0 was used for data analysis.
RESULTS
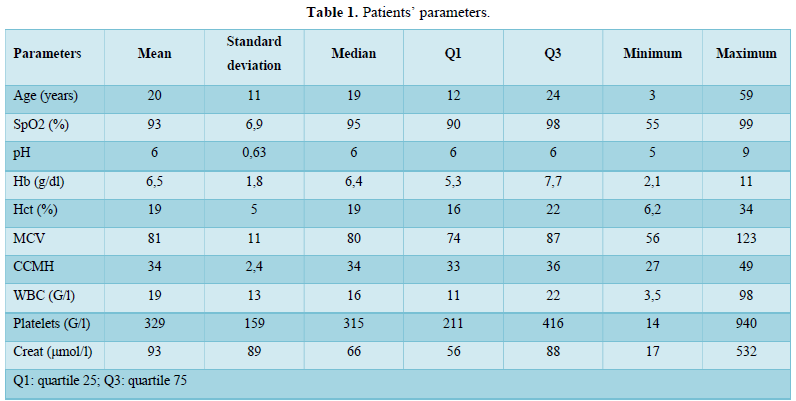
We included 219 patients in our study. There were 127 female patients (58%) and 92 male patients (42%). The sex ratio was 0.42. The mean age of the patients was 20 ± 11 years with extremes of 3 and 59 years. The most represented age group was 15-59 years (67.6%). Of these 219 patients, 136 were being followed up (62.9%); and 83 were not (37.1%). 108 patients (49.3%) had an acute hemolysis crisis; 106 patients (48.4%) had a Vaso occlusive crisis (bone or abdominal). Fever was found in 32.4% of cases. Malaria was the most common etiology. The hematological parameters are shown in Table 1. 61 patients (27.9%) had positive proteinuria; 48 patients (21.9%) had positive hematuria; 26 (11.9%) had positive leukocyturia; 7 (3.2%) had positive UD nitrites (Table 1). Only 47 patients had achieved creatinine levels and the mean was 93± 89 µmol/l with extremes of 17 and 532 µmol/l. Acute attacks of hemolysis were the most frequent emergency encountered, in 49.3% of cases (108 patients) (Table 2).

DISCUSSION
Renal manifestations in sickle cell disease are secondary to sickle cell disease, which is promoted and maintained by acidosis and hypoxia in the renal medullary [8,14]. Intracapillary sickle cell disease increases the blood viscosity of the renal medullary. This results in the formation of micro-thrombosis of the vasa recta, reactive secretion of vasodilating substances and deposition of interstitial hemosiderin. These phenomena are responsible for functional disorders, glomerular hyperfiltration, hematuria, renal infarction, papillary necrosis and interstitial fibrosis that can progress to chronic renal failure [14,15]. The lack of perfusion may lead to increased sensitivity of the renal parenchyma to infectious agents [8].
Study population
Our study population was predominantly female. The sex ratio was 0.42 in favor of women. This result is similar to those of other studies [16-19] which reported ratios ranging from 0.33 to 1.1 in favor of females. The study by Kaze [20] found a ratio in favor of men. This difference could be explained by the small sample size of their study which was 72 (one third of our sample size). The mean age of our patients, 20 ± 11 years, was identical to those found in the literature by many authors and ranged from 19.4 to 23.5 years [16,19-21]. This average age is slightly lower than that of the Malian series by Fongoro [9], which was 30.1 years. This result reflects an increase in the life expectancy of our sickle cell patients over the last few years, an increase that would be linked to the improvement of their management.
Proteinuria (UP)
This is the first urinary anomaly found in our study in terms of frequency. It was found in 27.9% of cases. This result of a positive UP at the UD is similar to those of other studies which reported values of between 20% and 54% in homozygous sickle cell patients [9,20,21]. This result is significantly higher than those reported by Nke Ateba [16] in Yaoundé, Ogwu [22] in Port Harcourt, Nigeria, Olorukooba [23] in Zaria, Nigeria, Uchenna [24] in Nigeria and Osei-Yeboah [25] in Accra, Ghana, which were respectively 10.8%, 7, 6.5%, 3.4% and 2.8%. This difference could be explained in the case of the Nke Ateba study by the inclusion criteria which eliminated any subject who was ill or had been transfused in the last three months before the study period. As for Ogwu, Olorukooba, Uchenna and Osei-Yeboah, their work focused only on pediatric populations (children aged 16 months to 16 years, 2 to 17 years, 2 to 18 years and 1 to 12 years respectively). It is therefore appropriate to recall that proteinuria and microalbuminuria do not appear before the age of 7 years and their prevalence increases with age [14,18,25-27]. This increase with age would explain the prevalence observed in samples consisting mainly of adults. This is in agreement with Ebah who emphasizes that proteinuria is related to the duration of sickle cell disease [11,28]. It is therefore important to test at least twice a year for dipstick UP or microalbuminuria, starting at the age of 5 years [7,22,25].
Hematuria (UH)
This is one of the most common manifestations in sickle cell disease (sickle cell nephropathy), regardless of age. It is almost always macroscopic and painless, but can also be microscopic and painful or not. Hematuria may originate in one or both kidneys. It results from papillary necrosis or micro thrombotic infarcts in the vasa-recta and peritubular capillaries of the renal medulla, with extravasation of blood into the collecting tubes [14,15,29]. It is also associated with infections or delayed transfusion reactions. Hematuria should be investigated systematically for urinary tract infection or papillary necrosis.
UH is the second most common renal event in our series. It was positive in 21.9% of our patients. This result is in line with those of many African series south of the Sahara which report frequencies ranging from 10.3% to 26% [17,20,23,30,31]. The frequencies of 11% [22], 6.7% [24], 5.6% [19] and 3.3% [25] reported respectively by Ogwu, Uchenna, Fall and Osei-Yeboah were lower than the 21.9% frequency found in our study. The small number of patients (36 in Fall's study; 60 in Uchenna’s study and 72 in Ogwu’s study) could justify this difference. The disparity in frequencies could also be explained by the fact that these studies are pediatric series, and that they were carried out in the inter-critical phase only. Also, possible errors in the reading of urine strips cannot be excluded.
Leukocyturia and nitrites
The increased susceptibility of sickle cell patients to urinary tract infections compared to the general population has been widely demonstrated in the literature.
Asymptomatic bacteriuria is common in sickle cell patients compared to the general population [25,32-36], and even more so in female sickle cell patients [35,36]. It can lead to overt urinary tract infection. In our series, leukocytes and nitrites, known to be indicators of a potential urinary tract infection, were found at frequencies of 11.9% and 3.2% respectively. These values are identical to those found by Osei-Yeboah in Accra, Ghana, who reported leukocyturia at 12.6% and 2.2% for nitrite in a pediatric series [25]. In this series, the controls with AA children were 5.7% and 1.4% respectively. Kaze [20] in Yaoundé, Cameroon, reported a frequency of leukocyturia of 77.8%. This difference could be explained by the small sample size of their study (72) compared to ours. Any asymptomatic urinary tract infection should therefore be identified and treated to prevent renal complications. The urine dipstick is a key means of early detection of these infections specially in a resource limited setting, in order to initiate treatment to minimize these complications.
Our study had limitations: creatinine levels in admitted patients and cytobacteriological examination of urine requested in patients with leukocyturia and/or nitrites at BU were only performed by a minority of patients. 24-hour proteinuria was not performed in any of the patients with positive UD proteinuria. Renal ultrasound was not routinely requested in patients with elevated creatinine levels. The main reason for these shortcomings is a financial problem.
CONCLUSION
Urinary anomalies are common in sickle cell disease. The urine dipstick is a simple and effective means of screening for sickle cell nephropathy. It should be performed systematically in all sickle cell patients, whether they are seen in an acute crisis or as part of a routine visit. This would allow early detection of nephropathy and the initiation of measures to avoid spontaneous progression to chronic renal failure, which is not easy to manage in our context.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
AUTHORS’ CONTRIBUTIONS
Data collection and entry, interpretation of results and writing of the article were carried out by Talomg Tamekué. Data analysis was carried out by Simo Louokdom. Tchidjo Ngamo, Malanda and Ngolet Ocini contributed to the writing of the article and its critique. Elira Dokékias supervised the work. The final version of the manuscript was read and approved by all authors.
ACKNOWLEDGEMENTS
We would like to thank the National Reference Centre for Sickle Cell Disease “Antoinette SASSOU NGUESSO” and its staff, as well as all the patients who consented to participate in this study.
- Rees DC, Williams TN, Gladwin MT (2010) Sickle-cell disease. Lancet 376: 2018-2023.
- Labie D, Elion J (2005) Molecular and pathophysiological basis of hemoglobin diseases. EMC-Hématologie 2: 220-239.
- Mongardon N, Habibi A, Vodovar D, Cherait C, Haouache H, et al. (2015) Acute sickle cell crisis. Le Congrès Médecins. Conférence d’Actualisation. pp: 1-16.
- Bouanani N, El Bakkouri J, Faez S, Benchemsi N (2013) Renal involvement in sickle cell disease patients. Les technologies de laboratoire. 8: 31.
- Ndiaye FSD, Fall S, Niang A, Diop S, Diouf B (2006) Chronic renal failure in homozygous sickle cell disease patients in Dakar. Rev Int Sci Méd 8(3): 23-26.
- Ocheke IE, Mohamed S, Okpe ES, Bode-TF, Mc Cullouch MI (2019) Microalbuminuria risks and glomerular filtration in children with sickle cell anemia in Nigeria. Ital J Pediatr 45: 143.
- Corinne P (2008) Renal surveillance in sickle cell disease. mt pédiatrie. 11(1): 47-52.
- Raynal G, Bracq A, Tillou X, Limani K, Petit J (2007) Renal complications of sickle cell disease. Progrès en Urologie 17: 794-795.
- Fongoro S, Diallo D, Diallo DA, Tchiango KA, Maiga MK (2009) Kidney diseases associated with the sickle cell gene in the nephrology and hemodialysis department of the CHU du point G. Mali Méd XXIV: 2.
- Kabir OO, Nwamaka DE, Sagar UN, Vela-Parada XF, Achebe MM, et al. (2019) Sickle Cell Nephropathy in the Pediatric Population. Blood Purif 47: 205-213.
- Sharpe CC, Thein SL (2011) Sickle cell nephropathy-a practical approach. Br J Haematol 155(3): 287-297.
- Ataga KI, Orringer EP (2000) Renal abnormalities in sickle cell disease. Am J Hematol 63: 201-211.
- Powars DR, Elliot-Mills DD, Chan L, Niland J, Hiti AL, et al. (1991) Chronic renal failure in sickle cell disease: Risk factors, clinical course and mortality. Ann Intern Med 115: 614-620.
- Scheinman JI (2009) Sickle cell nephropathy. In Pediatric Nephrology, Avner ED, Harmon WE, Niaudet P, Yoshikawa N. (eds.), 6th ed,. Berlin, Heidelberg. Springer Vol: 2.
- Saborio P, Scheinman JI (1999) Sickle cell nephropathy. J Am Soc Nephrol 10: 187-192.
- Nke Ateba G, Ngo Sack FF, Ateba MHG, Ngongang J (2017) Exploration of Glomerular Function in Homozygous Sickle Cell Patients in Yaoundé. Health Sci Dis 18: 2.
- Ademola AE, Olanrewaju TA (2013) Correlation between dipstick urinalysis and urine sediment microscopy in detecting hematuria among children with sickle cell anemia in steady state in Ilorin, Nigeria. Pan Afr Med J 15: 135.
- Iwalokum BA, Iwalokum SO, Hodomu SO, Aina OA, Agomo PU (2012) Evaluation of microalbuminuria in relation to asymptomaticbacturia in Nigeria patients with sickle cell disease. Saudi J Kidney Dis Transpl 23(6): 1320-1330.
- Fall S, Seck SM, Cissé MM (2010) Les atteintes rénales associées à la drépanocytose homozygote SS à Dakar. Dakar Med 55: 3.
- Kaze FF, Kengne AP, Atanga LC, Lobe MM, Menanga AP, Halle MP, et al. (2013) Kidney function, urinalysis abnormalities and correlates in equatorial Africans with sickle cell disease. Clin Kidney J 6: 15-20.
- Abdu A, Emokpae MA, Uadia PO, Kuliya-Gwarzo A (2011) Proteinuria among adult sickle cell anemia patients in Nigeria. Ann Afr Med 10(1): 34-37.
- Ugwu RO, Eke FU (2007) Urinary abnormalities in children with SCA. Port Harcourt Med J 2: 45-50.
- Olorukooba AA, Akuse RM, Ogunrinde GO, Mamman AI, Yusuf R, et al. (2018) Renal abnormalities among children with sickle cell anemia. 45(2): 112-117.
- Uchenna MN, Chukwukere OC, Uche OH, Achigbu KI (2020) Sickle Cell Nephropathy and Associated Factors among Asymptomatic Children with Sickle Cell Anemia. Int J Pediatr 2020: 1286432.
- Osei-Yeboah CT, Rodrigues O (2011) Renal status of children with sickle cell disease in Accra, Ghana. Ghana Med J 45: 155-160.
- Emokpae MA, Uadia PO (2012) Sickle cell disease and renal disease, Diseases of Renal Parenchyma.
- Revuelta KL, Andrès RMP (2011) Kidney abnormalities in sickle cell disease. Nefrologia 31(5): 591-601.
- Leonard EM (2013) Renal involvement in sickle cell disease: An African perspective for an African condition. Clin Kidney J 6: 6-7.
- Pham PT, Pham PC, Willinson AH, Lew SQ (2000) Renal abnormalities in sickle cell disease. Kidney Int 57(1): 1-8.
- Sesso R, Almeida MA, Fitgueiredo MS, Bordin JO (1998) Renal dysfunction in patients with sickle cell anemia or sickle cell trait. Braz J Med Biol Res 31: 1257-1262.
- Ugwu R, Eke F (2007) Urinary abnormalities in children with sickle anemia. Port Harcourt Med J 2: 45-50.
- Gausch A, Cua M, Mitch WE (1996) Early detection and the course of glomerular injury in patients with sickle cell anemia. Kidney Int 49: 786-791.
- Cumming V, Ali S, Forrester T, Roye-Green K, Reid M (2006) Asymptomatic bacteriuria in sickle cell disease: A cross sectional study. BMC Infect Dis 6: 46.
- Asinobi AO, Fatunde OJ, Brown BJ, Osinusi K, Fasina NA (2003) Urinary tract infection in febrile children with sickle cell anemia in Ibadan, Nigeria. Ann Trop Paediatr 23(2): 129-134.
- Donkor SE, Osei AJ, Anim-Baidoo I, Darkwah S (2017) Risk of Asymptomatic Bacteriuria among People with Sickle Cell Disease in Accra, Ghana. Diseases 5(1): 4.
- Chukwu BF, Okafor HU, Ikefuna AN (2011) Asymptomatic bacteriuria in children with sickle cell anemia at The University of Nigeria teaching hospital, Enugu, South East, Nigeria. Ital J Pediatr 37: 45.
-
 Table 1
Table 1 -
Table 2


QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Cancer Science and Treatment (ISSN:2641-7472)
- International Journal of Diabetes (ISSN: 2644-3031)
- Chemotherapy Research Journal (ISSN:2642-0236)
- Journal of Nursing and Occupational Health (ISSN: 2640-0845)
- Journal of Psychiatry and Psychology Research (ISSN:2640-6136)
- Journal of Infectious Diseases and Research (ISSN: 2688-6537)
- Journal of Rheumatology Research (ISSN:2641-6999)


