Journal of Pharmaceutics and Drug Research (ISSN:2640-6152)

Review Article
Review on Importance of Computer Aided Drug Design in Drug Discovery
6279
Views & Citations5279
Likes & Shares
CADD (computer aided drug design) is a technique which uses softwares for predicting the structure, value of properties of known, unknown, stable and molecular species using mathematical equations.the various method includes are Molecular modelling, molecular mechanics, molecular docking, quntam mechanics, hybrid QM/MM,QSAR and perform detail in molecular docking studies. It is estimated that a typical drug discovery cycle, from lead identification through to clinical trials, can take 14 years with cost of 800 million US dollars. Addition of computer aided drug design technologies to the R&D approaches of a company, could lead to a reduction in the cost of drug design and development by up to 50%.
Keywords: Computer aided drug design, Molecular modelling, Molecular mechanics, Molecular docking, Quntam mechanics, Hybrid QM/MM, QSAR
INTRODUCTION
Computer Aided Drug Design (CADD)
CADD (computer aided drug design) is a technique which uses softwares for predicting the structure value of properties of known, unknown, stable and unstable molecular species using mathematical equations [1,2].
Computers are essential tool in modern chemistry and are important in both drug discovery and development. CADD is used in pharmaceutical industry to discover, design and optimize new, effective in safe drugs [3].
Why CADD?
- i) The time consumption and financial investment involved in drug discovery is reduced by the utilization of computer aided drug design [4].
It is estimated that a typical drug discovery cycle from lead identification to clinical trials can take 14 years with cost of 800 million us dollars. Addition of computer aided drug design technologies to the R&D approaches of a company could lead to a reduction in the cost of drug design and development by up to 50% [5].
ii)The medicinal chemist evaluate interaction between a compound and target site before synthesizing that compound, this means that, medicinal chemist need only synthesis and test the compounds that considerably increase the potency there by the chance of discovery of a potent drug is increased and thereby reducing that cost of drug development [6].
iii) There is an ever-growing effort to apply computational power to the combined chemical and biological space in order to streamline drug discovery, design, development and optimization [7].
METHODOLOGY
The more recent foundations of CADD were established in the early 1970s with the use of structural biology to modify the biological activity of insulin 6 and to guide the synthesis of human hemoglobin ligands. At that time, X-ray crystallography was expensive and time-consuming, rendering it infeasible for large-scale screening in industrial laboratories. Over the years, new technologies such as comparative modeling based on natural structural homologues have emerged and began to be exploited in lead design. These together with advances in combinatorial chemistry, high-throughput screening technologies and computational infrastructures, have rapidly bridged the gap between theoretical modeling and medicinal chemistry. Numerous successes of designed drugs were reported, including dorzolamide for the treatment of cystoid macular edema, zanamivir for therapeutic or prophylactic treatment of influenza infection, Sildenafil for the treatment of male erectile dysfunction, and amprenavir for the treatment of HIV infection [8].
VARIOUS MODELS
Molecular modelling
Molecular modeling is a general term that covers a wide range of molecular graphics and computational chemistry techniques used to build, display, manipulate, simulate and analyze molecular structure and to calculate properties of these structures.
To a chemical physicist, molecular modeling implies performing a high-quality quantum Mechanical calculation using a super computer on the structure; to a medicinal chemist, molecular modeling mean displaying and modifying a candidate drug molecule on the desktop computer. molecular modeling techniques can be divided into molecular graphics and computational chemistry [9].
Molecular graphics
Molecular graphics is the core of a modeling system, providing for the visualization of molecular structure and its properties. In molecular modeling; the data produced are converted into visual image on the computer screen by graphic packages. These images may be displayed in a variety of styles like space fill, CPK (Corey-Pauling-Koltum), stick, mesh, ball and stick and ribbon and colour scheme with visual aids. Ribbon presentation is used for larger molecules like nucleic acids and proteins (Figure 1).
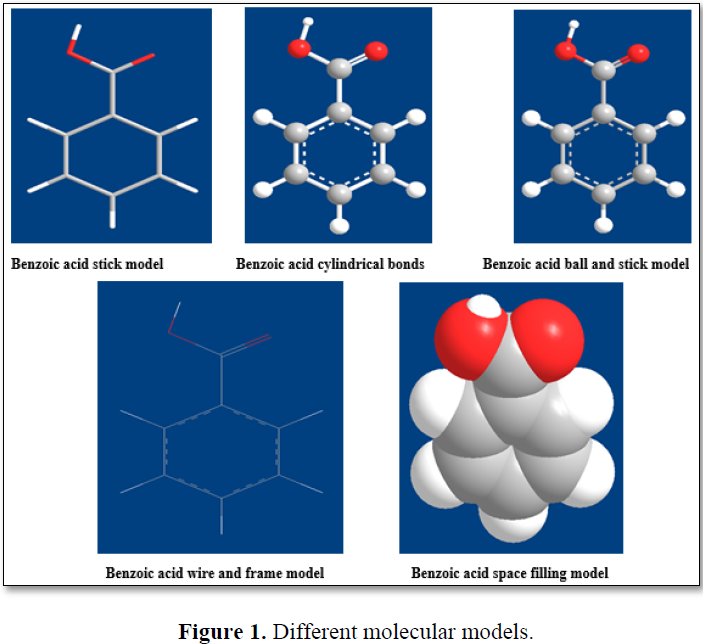
Visualization of molecular properties is an extremely important aspect of molecular modeling. The properties might be calculated using a computational chemistry program and visualized as 3D contours along with the associated structure. The most common computational methods are based on either molecular or quantum mechanics. Both these approaches produce equation for the total energy value for the structure. In these equations the position of the atom in the structures are represented by either Cartesian or polar co-ordinates. Once the energy equation is established, the computer computes a set of co-ordinates which corresponds to minimum total energy value for the system. This set of co-ordinates is converted into the required visual display by the graphic packages. The program usually indicates the 3D nature of the molecule and it can be viewed from different angles and allows the structure to be fitted to its target site. In addition, it is also possible by molecular dynamics, to show how the shape of structure might vary with time by visualizing the natural vibratration of the molecule [10].
Molecular computational chemistry
Molecular properties of a particular structure can be calculated by using computational chemistry programs. Computational chemistry is based on either molecular mechanics or quantum mechanics.
Molecular mechanics
Molecular mechanics is based on the assumption that the position of nuclei of the atom forming the structure is determined by the force of attraction/ repulsion operating in that structure.
Molecules are treated as a series of sphere (atoms) connected by spring (the bond) using this Model: E total is expressed mathematically by equation known as force fields given by:
E total=∑ E stretching + ∑ E bend + ∑ E torsion + ∑ E vdw + ∑E Columbic.
E stretching: it is the bond stretching energy.
E bend: is the bond energy
E torsion: is the torsional energy
E vdw: is the total energy contribution due to Vander Waal’s force
E columbic: the electrostatic attractive and repulsive forces operating in the molecule between the atoms carrying a partial or full charge.
Modeling can be created by utilizing molecular mechanics by using the following:
Commercial force filed computer program: Commercial packages usually have several different force fields with in the same package and it is necessary to pick the most appropriate one for the structure being modeled.
Assembling model: Molecular models are created by assembling a model from structural fragments held in the data base of the molecular modeling program .initially, these fragments are put together in a reasonably sensible manner to give a structure that does not allow for steric hindrance it is necessary to check that, the computer has selected atoms for the structure whose configuration corresponds to the type of bond required in structure.
The molecular mechanic methods require less computing time than the quantum mechanical approach and may be used for large molecules containing more than 1000 atoms [11].
Molecular dynamics
Molecular dynamics show the dynamic nature of the molecule by stimulating the natural motion of the atom in a structure.
Molecular dynamics can also be used to find minimal energy structure and conformational analysis.
Conformational analysis: Using molecular mechanics, it is possible to generate a variety of different conformations by using a molecular dynamics program which ˈheatsˈ the molecule to 800-900k .it means that the program allows the structure to undergo bond stretching and bond rotations as if it being heated at 800-900k.
In this process, the molecule is heated at high T (900k) for a certain period, and then cooled to 300k. To give the final structure, the process can be repeated, many different structures as required. Each of these structures can then be recovered, energy minimized and its steric energy measured. Out of this procedure, it is usually possible to identify distinct conformations, some of which might be more stable than the initial conformation [12].
Molecular docking
This technique is able to predict possible orientations of one molecule to another.
Molecular docking comprises of three consecutive steps:
- i) Defining of the binding site,
- ii) Placement of the ligand inside the defined site
iii) Evaluation of this placement called scoring.
Docking represents ligand binding to its receptor or target protein.
Docking is used to identify and optimize drug candidates by examining and modeling molecular interactions between ligand and target macromolecules. In docking multiple ligand conformations and orientations are generated & the most appropriate ones are selected [13].
How docking is done?
Preparation of molecule: The molecule benzoic acid is drawn in chem draw ultra-software and then molecule is converted into pdb format (protein data bank) after minimization of energy (e.g: benzoic acid 0.94 kcal/mol) (Figure 2).
Preparation of proteins: A protein (HVR protease) is selected from pdb and it is imported in to MVD and the cavities are identified followed by docking with the ligand (benzoic acid).
Protein docking: Docking is performed in MVD to obtain the molecular dock score. Higher the score and higher the hydrogen bond length, greater is the affinity for the compound towards that protein [14].
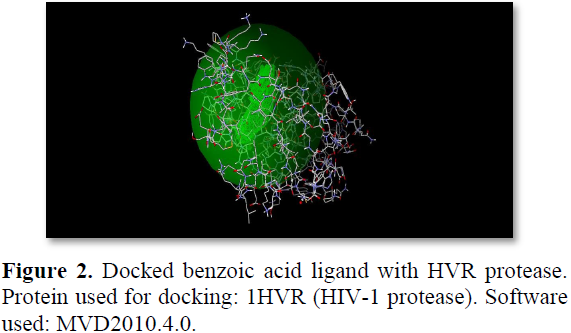
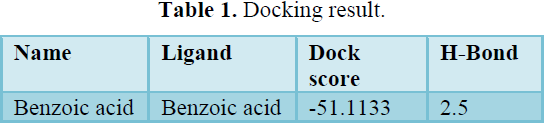
Docking result (Table 1)
Scoring methods are used to rank the affinity of ligands to bind to the active site of a receptor. In virtual high throughput screening compounds are docked into an active site and then scored to determine which once more likely to bind tightly to the target macromolecule.
Quantum mechanics
It’s based on the realization that electrons and their material particles exhibit wave like properties. This allows the well defined, parameter free, mathematics of wave motion to be applied to electrons, atomic and molecular structure. The basis of these calculations is the Schrödinger wave equation, which in its simplest form may be stated as:
where,
Eⱷ -total potential and kinetic energy of all the particles in the structure
H - Hamiltonian operator acting on the wave function ⱷ.
Quantum mechanical methods are suitable for calculating the following:
- Molecular orbital energies and coefficients.
- Heat of formation for specific conformations.
- Partial atomic charges calculated from molecular orbital coefficients.
- Electrostatic potential.
- Dipole moments.
- Transition state geometrics and energies.
- Bond dissociation energies [15].
QSAR (Quantitative structural activity relationship)
QSAR provides a wide variety of descriptors that you can use in determining new QSAR relationships [16].
HYBRID QM∕MM
Quantum mechanical methods are very powerful however they are computationally expensive, while the molecular mechanical methods are fast but suffer from several limitations. A new class of method has emerged that combine the good points of QM (accuracy) and MM (speed) calculations. These methods are known as mixed or hybrid quantum–mechanical and molecular mechanics method (hybrid MM or QM). The methodology for such techniques was introduced by another study [17].
Energy Minimization
It is also called energy optimization or geometry optimization; it is used to compute the equilibrium configuration of molecules and solids. By this technique we can only obtain a final state of system that corresponds to minimum of potential energy. In Energy minimization one can obtain a molecule with least energy state i.e. zero energy state. In this state molecule get equilibrium configuration. Energy minimization tools are GAMESS Ghemical PS13 TINKER Ghemical can be used or PS13 for quantum mechanical calculations. If proteins are used, a program such as PyMol can be used to identify ligand binding pockets, together with the Deep View PDB viewer to investigate the amino acid sequences of the protein. To transfer files between programs, Open Babel might be useful or even required to interconvert the file formats [18].
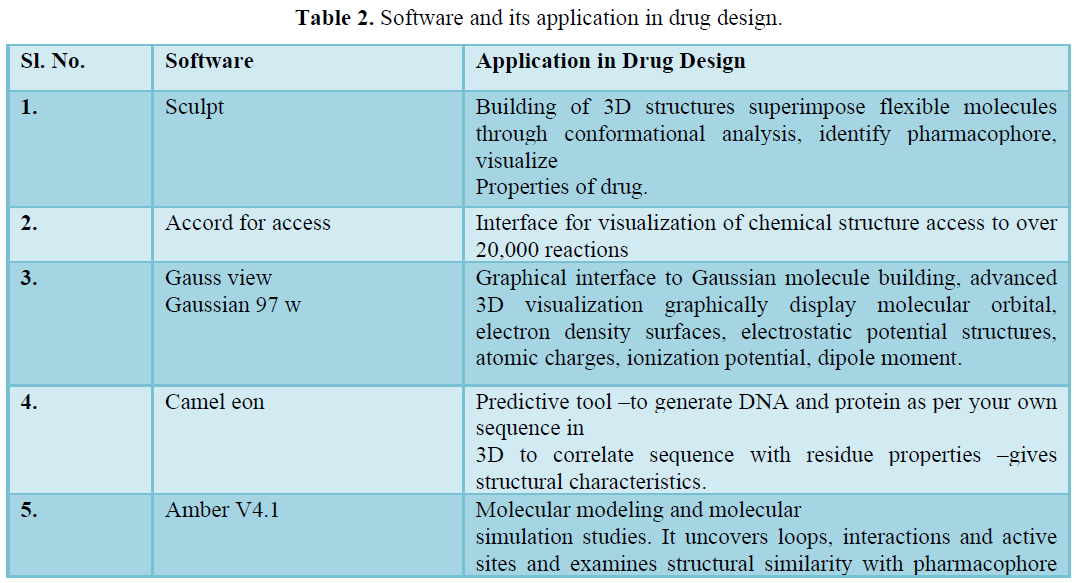
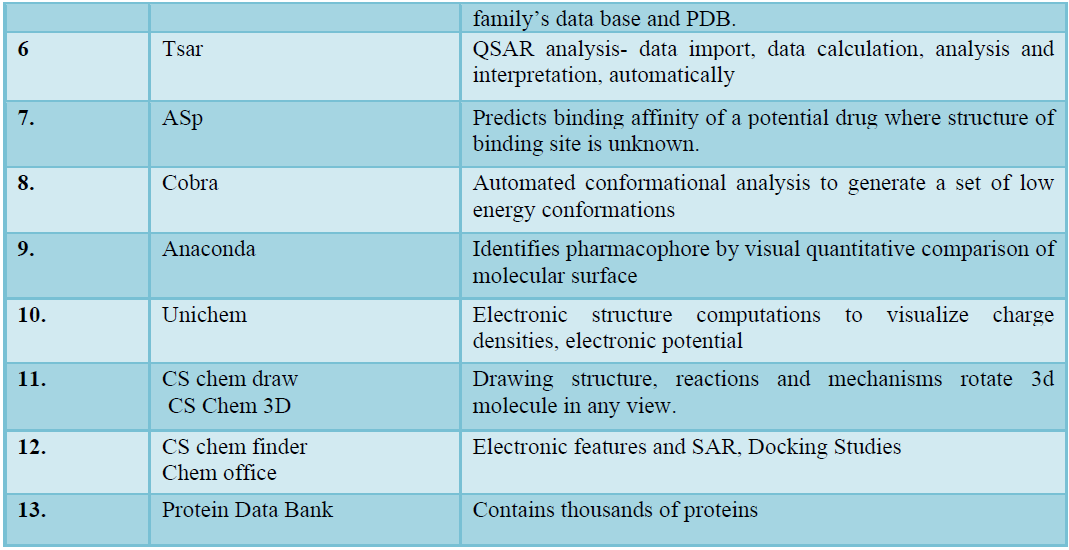
SOFTWARE’S AND ITS APPLICATION IN DRUG DESIGN (TABLE 2)
LIMITATION OF COMPUTER AIDED DRUG DESIGN
Lengthy, expensive and requires technical expertise.
CONCLUSION
CADD is now widely recognized as a viable alternative and complement to high throughput screening. The search for new molecular entities has led to the construction of high-quality datasets and design libraries that may be optimized for molecular diversity or similarity. Conversely, advances in molecular docking algorithms, combined with improvements in computational infrastructure, are enabling rapid improvement in screening throughput. Propelled by increasingly powerful technology, distributed computing is gaining popularity for large-scale screening initiatives. Combined with concerted efforts towards the design of more detailed physical models such as solubility and protein solvation, these advancements will, for the first time, allow the realization of the full potential of lead discovery by design.
FUTURE PROSPECTIVE
- Development and application of computational techniques for prediction of free energies of binding of drugs.
- Development and application of new methods for carbohydrate computational chemistry.
- Biomolecular simulation studies of proteins, sugars and DNA.
- QM/MM studies of the condensed phase.
- Molecular modelling to obtain 3-dimensional structures for proteins we are interested in as targets for drug design, to design mutations or to study potential interactions with other proteins or nucleic acids.
- Designing lead drug structures and molecules which bind to enzyme active sites, to DNA or to tRNA or ribozymes.
- Re-designing proteins for molecular engineering, for example to produce variants of Green Fluorescent Protein that can be specifically chemically labelled to register enzyme action, such as the action of caspase 3 inside cells undergoing apoptosis.
- Designing molecules with novel chemical activities such as DNA cleavage.
- Understanding the conformational properties and energetics of small molecules.
- Determining high-resolution structures of chemically modified nucleic acids or of DNA: drug complexes using full distance geometry restraints combined with high- field NMR structural determinations of nucleic acid structures.
- Understanding how different families of ligands dock into binding sites of macromolecules.
- Springer Rational Drug Design (1999) Verlag: New York, pp: 171-89.
- Nag A, Dey B (2011) Computer-aided drug design and delivery systems. The McGraw-Hill Companies, Inc: Mumbai, pp: 255-301.
- Banker G (2005) Computer-aided Drug Design. Marcal Decker Inc: New York, pp: 7-48.
- Weiling P (2009) Guidebook on molecular modeling in drug design. Academic Press: Oxford, London, pp: 13-56.
- Williams HH (1996) Reviews in Computational Chemistry. VCH Publishers: New Delhi, pp: 19-42.
- Baldi A (2010) Computational approaches for drug design and discovery: An overview. Sys Rev Pharm 1(1).
- Whittle PJ, Blundell TL (1994) Protein structure-based drug design. Annu Rev Biophys Biomol Struct 23: 349-375.
- Itzstein MV, Wu WY, Kok GB (1993) Rational design of potent sialidase-basedinhibitors of influenza virus replication. Nature 363: 418-423.
- Valentina IK (2010) Molecular modeling. Chem Eur J 2(2): 422-424.
- Ilango KV (2010) Molecular graphics. Der Pharma Chemica 2(2): 424-426.
- Duprat AF, Huynh T, Dreyfus G (1998) Toward a principled methodology for neuralnetwork design and performance evaluation in QSAR: Application to the prediction of Log P. J Chem Inf Comput Sci 38(4): 586-594.
- Clare BW, Supuran CT (2005) A physically interpretable quantum-theoretic QSAR for some carbonic anhydrase inhibitors with diverse aromatic rings, obtained by a new QSAR procedure. Bioorg Med Chem 13(6): 2197-2211
- Perdo, Lei H (2010) A systematic review on CADD: Docking and Scoring. JMPI, pp: 47-51.
- Yang JM, Chen CC (2004) GEMDOCK: A generic evolutionary method for molecular docking. Proteins 55: 288-304.
- Clare BW, Supuran CT (2004) Quantum theoretic QSAR of benzene derivatives: Some enzyme inhibitors. J Enz Inhib Med Chem 19: 237-248.
- Clare BW, Supuran CT (2005) Predictive flip regression: A technique for QSAR of derivatives of symmetric molecules .45: 1385-1391.
- Tetko IV (2002) Application of associative neural networks for prediction of lipophilicity in ALOGPS 2002, pp: 1136-1145.
- Downs, GM Bultinck (2004) Computational Medicinal Chemistry for Drug Discovery. Marcel Dekker, New York 2004, pp: 515-538.
-
 Table 1
Table 1 -
Table 2
-
Table 3







