2593
Views & Citations1593
Likes & Shares
Nicotinamide is a water-soluble amide form of niacin (nicotinic acid or vitamin B3). Both niacin and nicotinamide are widely available in plant and animal foods and niacin can also be synthesized in the liver from dietary tryptophan. Nicotinamide is also commercially available in vitamin supplements and in a range of cosmetic, hair and skin preparations. Nicotinamide is the primary precursor of nicotinamide adenine dinucleotide (NAD+), an essential coenzyme in ATP production and the sole substrate of the nuclear enzyme poly-ADP-ribose polymerase-1 (PARP-1). Numerous in vitro and in vivo studies have clearly shown that PARP-1 and NAD+ status influence cellular responses to genotoxicity which can lead to mutagenesis and cancer formation.
Keywords: Nicotinamide, NAD+ levels, Nuclear enzyme poly-ADP-ribose polymerase-1, Niacin, Pyridine-carboxylic acids
INTRODUCTION
Niacin, nicotinamide and cancer
Niacin, also known as nicotinic acid, is an organic compound and is, depending on the definition used, one of the 20 to 80 essential human nutrients. Together with nicotinamide it makes up the group known as vitamin B3 complex [1]. It has the formula C6H5NO2 and belongs to the group of the pyridine-carboxylic acids. Nicotinamide may be obtained from the diet where it is present primarily as NAD+ and NADP+. These are hydrolysed in the intestine and the resulting nicotinamide is absorbed either as such, or following its hydrolysis to nicotinic acid. Nicotinamide is present in nature in only small amounts. In unprepared foods, niacin is present mainly in the form of the cellular pyridine nucleotides NAD and NADP. Enzymatic hydrolysis of the co-enzymes can occur during the course of food preparation. Boiling releases most of the total niacin present in sweet corn as nicotinamide (up to 55 mg/kg). Nicotinamide may be toxic to the liver at doses exceeding 3 g/day for adults [2].
The prime cause of cancer is the damage to the mitochondria in normal cells. Nearly all cancer cells contain damaged mitochondria and the basic reason behind this, is increasing the intracellular inflammation or basically the incline in Reactive Oxygen Species (ROS) produced by each mitochondrion in oxidative phosphorylation. Increasing the ROS in a cell can cause damage to the DNA of the mitochondrion and also nucleus DNA, but another reason behind turning the normal cell into cancer cell is the chaos caused by the increasing of inflammation inside each cell and increasing the intracellular ROS. These chaos causes some abnormal messaging between the DNA of the nucleus to stop the apoptosis and turning the oxidative phosphorylation to the fermentation in cytosol. Normally by damaging to the mitochondria, the cell should apoptosis. However; the nucleus sends wrong messages to stop the apoptosis and do fermentation process in cytosol to survive the cell. Even some normal left mitochondria would be shut down and stop the oxidative phosphorylation. This is the main and the real reason how increasing intracellular inflammation can cause cancer. This research introduces the butterfly effect inside the normal cells is the basic reason behind the cause of cancer [3].
MATERIALS & METHODS
There are relatively few epidemiological studies on the association between nicotinamide intake and cancer in humans. Deficiency of nicotinamide and other micronutrients including riboflavin, zinc and magnesium have been linked to the increased frequency of oesophageal cancer in certain populations in China and Italy [4, 5]. Low dietary niacin has also been associated with an increased frequency of oral, gastric, and colon cancers, as well as oesophageal dysplasia [6-8].
In the Linxian trial in China, involving nearly 30,000 residents, 40 mg niacin and 3.2 mg riboflavin were supplemented in one of the treatment arms daily for over 5 years. It was shown that this combined supplementation decreased oesophageal cancer incidence and mortality by 14% and 10%, respectively [9]. Most human studies have examined the dietary intake or supplementation of niacin in combination with other micronutrients [10,11].
The impact of niacin on human carcinogenesis is therefore confounded by the effect of other micronutrients. Analysis from a large Western population within The Malm¨o Diet and Cancer Study in Sweden showed that approximately 15%-20% of individuals in this population were niacin deficient [12]. While severe niacin deficiency resulting in pellagra is uncommon in Western populations, suboptimal niacin intake may be relevant in populations at risk such as cancer patients and individuals with high occupational or environmental exposure to genotoxic agents including ionizing radiation, ultraviolet radiation (UVR) and alkylating agents. Limited studies indicate that cancer patients are at risk of niacin deficiency [13,14].
In one trial involving 42 patients with various primary cancers, it was shown that 40% of these patients were niacin deficient as measured by abnormally low urine levels of the niacin metabolite N1methyl nicotinamide [15]. Chemotherapy may also depress NAD+ levels [16,9] and precipitate pellagra by promoting anorexia and malabsorption. Some chemotherapeutic agents (e.g., 5-fluorouracil, 6-mercaptopurine) also interfere with tryptophan conversion to niacin [16]. Moreover, chemotherapeutic alkylating agents have been shown to cause miscoding lesions, chromosomal aberrations [17] and secondary cancer, particularly leukemia, which complicates chemotherapy in 10%-15% of cancer survivals [18]. More direct evidence comes from studies in rats, which showed that niacin deficiency significantly increases the risk of chemotherapeutic-induced secondary leukemia [19]. Niacin and NAD+ levels are important determinants of genomic response stogen otoxicinsults [17,4]. Maintaining an optimum nicotinamide level is therefore essential in cancer patients and individuals at risk of exposure to genotoxic agents [20].
Nicotinamide, which is the dietary precursor for NAD+, provides a substrate for PARP-1 activity. The activation of nuclear enzyme PARP-1 by DNA strand breaks during cellular genotoxic stress responses leads to complex signaling pathway that can enhance DNA repair, result in apoptotic cell death, or cause cellular energy loss leading to necrotic cell death. In vivo and in vitro studies showed that NAD+ content of the cells influences responses to DNA damaging agents. NAD+ depletion impairs ADP-ribose polymer metabolism and increases genomic instability in the face of genotoxic and oxidative stress challenges. Nicotinamide deficiency in humans may also contribute to increased frequency of gastrointestinal cancers in certain populations although other micronutrient deficiencies are likely to be involved as well. Nicotinamide supplementation in animal models has opposing effect on carcinogenesis, depending on the type of carcinogens and target organs. Nicotinamide protected against UV-induced immunosuppression in mice and humans and UV-induced carcinogenesis in mice. Limited study in humans indicates that skin NAD+ content is an important determinant of malignant phenotype. Thus, nicotinamide supplementation may influence the progression of premalignant actinic keratoses to malignant squamous cell cancers. PARP-1 plays a key role in regulation of genes involved in inflammation, apoptosis and cellular differentiation. While PARP-1 inhibition could impair its role in DNA repair, PARP-1 over-activation is detrimental to the cells by depleting its substrate NAD+, which leads to cellular energy crisis and necrotic cell death. In various murine models, PARP-1 inhibition was shown to favor apoptotic cell death, reduce inflammatory response and reduce genomic sensitivity to various carcinogens. However, extrapolation of these data to human, particularly when physiological regimes involved in human carcinogenesis, should be done cautiously. Further studies are needed to determine the effect of high-dose nicotinamide on in vivo carcinogenesis and genomic stability of the cancer cells and the surrounding normal cells.
There is a shortage of knowledge on the impact of niacin on cancer risk in human populations. It is known that cancer patients tend to be deficient in niacin at a time when they are exposed to large doses of genotoxic drugs during chemotherapy [21,22]. Between 5% and 10% of surviving chemotherapy patients develop secondary cancers, especially leukemias [23].
Although animal models suggest that niacin deficiency enhances this risk [24], there are no human data available to further define this risk. Although developed countries generally supplement niacin in cereal products, there may still be a significant proportion of these populations experiencing subclinical niacin deficiency [25-27]. Niacin and riboflavin were supplemented in one of the treatment arms of the Linxian trials in China [28]. Although these supplements did not provide any benefit to the oral/esophageal cancers in this study, there are various ways to interpret these results. The duration of the study was probably too short to examine the role of niacin during cancer initiation. In addition, the high esophageal cancer incidence in this population is associated with heavy contamination by fumonisin mycotoxins [29], which appear to promote carcinogenesis by a rather unique mechanism that may not be responsive to niacin status [30].
Human patients with hypercholesterolemia often are treated with high doses of nicotinic acid (greater than or equal to 3 g/day). Studies on the impact of these treatments on cancer incidence are few in number, cover relatively short periods of time and represent late stages in the carcinogenic process. However, nicotinic acid therapy did not seem to cause the small increase in cancer incidence observed in populations using a variety of other non-statin drugs to lower blood cholesterol [31].
Interestingly, nicotinic acid use for 6 years by patients with cardiovascular disease led to a decrease in all-cause mortality measured 8 years after the drug use was discontinued [32].
Epidemiological studies about niacin status and human cancer incidence often find significant associations, but interpretation of these is also difficult. In a variety of countries, including Iran, Africa, Italy, Switzerland and the United States, maize (corn) consumption (which causes niacin deficiency), or low levels of estimated niacin intake, have been associated with an increased frequency of gastric, colon, oral, or esophageal cancers [33-37].
This is interesting from the perspective that niacin deficiency may target the gastrointestinal tract, as evidenced by diarrhea during pellagra, likely caused by the rapid cell turnover of these tissues. Niacin deficiency also causes inflammation and hyperplasia in the esophagus [38], which would also promote cancer at this site. However, these experiments suffer from covariance between maize consumption and fumonisin exposure, and/or niacin status and fruit and vegetable consumption. No work has been done on the epidemiology of niacin deficiency and skin cancer, although animal models of niacin deficiency [39] and human familial DNA repair defects that mimic the sun sensitivity of pellagra [40] are associated with an increase in skin cancer risk [41].protoporphyrin IX ring. Impaired conversion of coproporphyrinogen III into protoporphyrin IX results in higher coproporphyrin excretion in urine. Moreover, it impairs ribonucleic acid metabolism in erythrocytes; damage to erythrocyte membrane through the inhibition of membrane ATPase leads to reduced blood cell survival and hemolysis. In the gastrointestinal tract, lead may damage the autonomic system, causing peristalsis abnormalities. The neurological toxicity of lead stems from the fact that it is highly lipid-soluble; poisoning results in degenerative changes in the cerebral cortex, the cerebellum and subcortical nuclei, and the hypothalamus autonomic centers as well as segmental demyelination of peripheral nerve fibers. After the absorption of a large amount of tetraethyl lead, patients present with acute poisoning; typically, there is a latent period, which may last between several hours and a few days and is followed by headache and dizziness, loss of appetite, insomnia and considerable weakness. A physical examination reveals decreased blood pressure and heart rate values. Patients show signs and symptoms of nervous system damage, such as paresthesia in the limbs, nystagmus and euphoria. Next, they develop mental disorders such as delirium, delusions, and sometimes schizophrenic syndrome. A period of agitation is followed by obtundation and sometimes death [20-25].
Toluene is a colorless liquid and has an odor similar to benzene. It may be absorbed by the lungs and skin. Toluene is metabolized to benzoic acid by methyl group oxidation and is excreted in urine within 15 h of the end of exposure. Symptoms of acute toluene poisoning include irritated mucous membranes, headache and dizziness, somnolence, and, rarely, loss of consciousness. Chronic poisoning is associated with pseudoneurotic disorders and possibly liver and kidney damage.
DISCUSSION
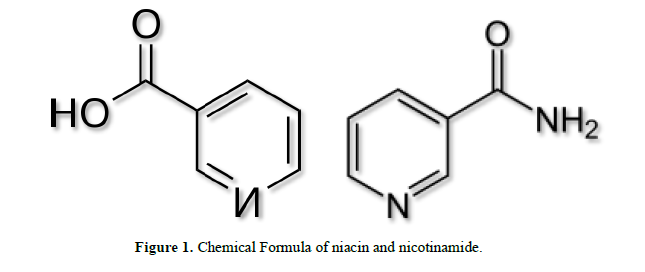
This colorless, water-soluble solid is a derivative of pyridine, with a carboxyl group (COOH) at the 3-position. Other forms of vitamin B3 include the corresponding amide nicotinamide, where the carboxyl group has been replaced by a carboxamide group (CONH2), as well as more complex amides and a variety of esters. Nicotinic acid and niacinamide are convertible to each other with steady world demand rising from 8,500 tons per year in the 1980s to 40,000 in recent years [42]. Niacin cannot be directly converted to nicotinamide, but both compounds are precursors of the coenzymes nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP) in vivo [43]. NAD converts to NADP by phosphorylation in the presence of the enzyme NAD+ kinase. NADP and NAD are coenzymes for many dehydrogenases, participating in many hydrogens transfer processes [44]. NAD is important in catabolism of fat, carbohydrate, protein and alcohol, as well as cell signaling and DNA repair and NADP mostly in anabolism reactions such as fatty acid and cholesterol synthesis [45]. High energy requirements (brain) or high turnover rate (gut, skin) organs are usually the most susceptible to their deficiency.
In animal models and in vitro, niacin produces marked anti-inflammatory effects in a variety of tissues-including the brain, gastrointestinal tract, skin and vascular tissue through the activation of NIACR1 [46-49]. Niacin has been shown to attenuate neuro-inflammation and may have efficacy in treating neuro-immune disorders such as multiple sclerosis and Parkinson's disease. Unlike niacin, nicotinamide does not activate NIACR1, however both niacin and nicotinamide activate the G protein-coupled estrogen receptor (GPER) in vitro [50].
The high doses of niacin used to improve the lipid profile have been shown to elevate blood sugar by 5-10%, thereby worsening diabetes mellitus [51]. Niacin therapy increases the risk of new-onset diabetes by approximately 34% [52].
CONCLUSION
There is a shortage of knowledge on the impact of niacin on cancer risk in human populations. Niacin deficiency may target the gastrointestinal tract, as evidenced by diarrhea during pellagra, likely caused by the rapid cell turnover of these tissues. Niacin deficiency also causes inflammation and hyperplasia in the esophagus which would also promote cancer at this site. No work has been done on the epidemiology of niacin deficiency and skin cancer, although animal models of niacin deficiency and human familial DNA repair defects that mimic the sun sensitivity of pellagra are associated with an increase in skin cancer risk. Maize consumption which causes niacin deficiency, have been associated with an increased frequency of gastric, colon, oral, or esophageal cancers. It is known that cancer patients tend to be deficient in niacin at a time when they are exposed to large doses of genotoxic drugs during chemotherapy and between 5% and 10% of surviving chemotherapy patients develop secondary cancers, especially leukemia. The biochemistry of vitamin B3 shows that high doses of Niacin increase the blood sugar levels in human and animals which can cause problems in cancer patients since cancer cells need glucose for respiration. For the treatment, it is better to use nicotinamide and niacin for the prevention of cancer.
1. Krutmann J, Humbert P (2011) Nutrition for healthy skin: Strategies for clinical and cosmetic practice. Springer Science Business Media.
2. Knip M, Douek IF, Moore WP, Gillmor HA, McLean AE, et al. (2000) Safety of high-dose nicotinamide: A review. Diabetologia 43: 1337-1345.
3. Zaminpira S, Niknamian S (2017) How butterfly effect or deterministic chaos theory in theoretical physics explains the main cause of cancer. EC Cancer 2.5: 227-238.
4. Blot WJ, Li JY, Taylor PR, Guo W, Dawsey S, et al. (1993) Nutrition intervention trials in Linxian, China: Supplementation with specific vitamin/mineral combinations, cancer incidence and disease specific mortality in the general population. J Natl Cancer Inst 85: 1483-1492.
5. Franceschi S, Bidoli E, Baron AE, La Vecchia C (1990) Maize and risk of cancers of the oral cavity, pharynx and esophagus in Northeastern Italy. J Natl Cancer Inst 82: 1407-1411.
6. Negri E, Franceschi S, Bosetti C, Levi F, Conti E, et al. (2000) Selected micronutrients and oral and pharyngeal cancer. Int J Cancer 86: 122-127.
7. Siassi F, Pouransari Z, Ghadirian P (2000) Nutrient intake and esophageal cancer in the caspian littoral of Iran: A casecontrol study. Cancer Detect Prev 24: 295-303.
8. Kabat GC, Miller AB, Jain M, Rohan TE (2008) Dietary intake of selected B vitamins in relation to risk of major cancers in women. Br J Cancer 99: 816-821.
9. Zablotska LB, Chen Y, Graziano JH, Parvez F, van Geen A, et al. (2008) Protective effects of B vitamins and antioxidants on the risk of arsenicrelated skin lesions in Bangladesh. Environ Health Perspect 116: 1056-1062.
10. Pelucchi C, Tramacere I, Bertuccio P, Tavani A, Negri E, et al. (2009) Dietary intake of selected micronutrients and gastric cancer risk: An Italian case-control study. Ann Oncol 20: 160-165.
11. Qu CX, Kamangar F, Fan JH, Yu B, Sun XD, et al. (2007) Chemoprevention of primary liver cancer: A randomized, double-blind trial in Linxian, China. J Natl Cancer Inst 99: 1240-1247.
12. Bosetti C, Scotti L, Maso LD, Talamini R, Montella M, et al. (2007) Micronutrients and the risk of renal cell cancer: A case-control study from Italy. Int J Cancer 120: 892-896.
13. Jacobson EL (1993) Niacin deficiency and cancer in women. J Am Coll Nutr 12: 412-416.
14. Inculet RI, Norton JA, Nichoalds GE, Maher MM, White DE, et al. (1987) Water-soluble vitamins in cancer patients on parenteral nutrition: Aa prospective study. J of Parenteral and Enteral Nutrition 11: 243-249.
15. Dreizen S, McCredie KB, Keating MJ, Andersson BS (1990) Nutritional deficiencies in patients receiving cancer chemotherapy. Postgraduate Medicine 87: 163-170.
16. Stevens HP, Ostlere LS, Begent RH, Dooley JS, Rustin MH (1993) Pellagra secondary to 5-fluorouracil. British J of Dermatology 128: 578-580.
17. Het Veld CWO, van Hees-Stuivenberg S, Zeeland AAV, Jansen JG (1997) Effect of nucleotide excision repair on hprt gene mutations in rodent cells exposed to DNA ethylating agents. Mutagenesis 12: 417-424.
18. Felix CA (1998) Secondary leukemias induced by topoisomerase targeted drugs. Biochim Biophys Acta 1400: 233-255.
19. Kirkland JB (2003) Niacin and carcinogenesis. Nutr Cancer 46: 110-118.
20. Toth B (1983) Lack of carcinogenicity of nicotinamide and isonicotinamide following lifelong administration to mice. Oncology 40: 72-75.
21. Negri E, Franceschi S, Bosetti C, Levi F, Conti E, et al. (2000) Selected micronutrients and oral and pharyngeal cancer. Int J Cancer 86: 122-127.
22. Siassi F, Pouransari Z, Ghadirian P (2000) Nutrient intake and esophageal cancer in the Caspian littoral of Iran: A case-control study. Cancer Detect Prev 24: 295-303.
23. Van Rensburg SJ, Bradshaw ES, Bradshaw D, Rose EF (1985) Oesophageal cancer in Zulu men, South Africa: A case-control study. Br J Cancer 51: 399-405.
24. Segal I, Hale M, Demetriou A, Mohamed AE (1990) Pathological effects of pellagra on the esophagus. Nutr Cancer 14: 233-238.
25. Shah GM, Le Rhun Y, Sutarjono I, Kirkland JB (2002) Niacin deficient SKH-1 mice are more susceptible to ultraviolet B radiation-induced skin carcinogenesis. Cancer Res 131: 3150.
26. Cleaver JE, Crowley E (2002) UV damage, DNA repair and skin carcinogenesis. Front Biosci 1.
27. Bryan GT (1986) The influence of niacin and nicotinamide on in vivo carcinogenesis. Adv Exp Med Biol 206: 331-338.
28. Toth B (1983) Lack of carcinogenicity of nicotinamide and isonicotinamide following lifelong administration to mice. Oncology 40: 72-75.
29. Rawling JM, Jackson TM, Roebuck BD, Poirier GG, Kirkland JB (1995) The effect of niacin deficiency on diethylnitrosamine-induced hepatic poly(ADP-ribose) levels and altered hepatic foci in the Fischer-344 rat. Nutr Cancer 24: 111-119.
30. Rawling JM, ApSimon MM, Kirkland JB (1996) Lung poly(ADP-ribose) and NAD+ concentrations during hyperoxia and niacin deficiency in the Fischer-344 rat. Free Radic Biol Med 20: 865-871.
31. Boyonoski AC, Gallacher LM, ApSimon MM, Jacobs RM, Shah GM, et al. (2000) Niacin deficiency in rats increases the severity of ethylnitrosourea-induced anemia and leukopenia. J Nutr 130: 1102-1107.
32. Boyonoski AC, Spronck JC, Jacobs RM, Shah GM, Poirier GG, et al. (2002) Pharmacological intakes of niacin increase bone marrow poly (ADP-ribose) and the latency of ethylnitrosourea-induced carcinogenesis in rats. J Nutr 132: 115-120.
33. Spronck JC, Kirkland JB (2002) Niacin deficiency increases spontaneous and etoposide-induced chromosomal instability in rat bone marrow cells in vivo. Mutat Res 508: 83-97.
34. Gensler HL, Williams T, Huang AC, Jacobson EL (1999) Oral niacin prevents photocarcinogenesis and photoimmunosuppression in mice. Nutr Cancer 34: 36-41.
35. Partida-Sanchez S, Randall TD, Lund FE (2003) Innate immunity is regulated by CD38, an ecto-enzyme with ADP-ribosyl cyclase activity. Microbes Infect 5: 49-58.
36. Berneburg M, Lehmann AR (2001) Xeroderma pigmentosum and related disorders: Defects in DNA repair and transcription. Adv Genet 43: 71-102.
37. Lautier D, Lagueux J, Thibodeau J, Menard L, Poirier GG (1993) Molecular and biochemical features of poly (ADP-ribose) metabolism. Mol Cell Biochem 122: 171-193.
38. Satoh MS, Poirier GG, Lindahl T (1993) NAD (+)-dependent repair of damaged DNA by human cell extracts. J Biol Chem 268: 5480-5487.
39. Shieh WM, Ame JC, Wilson MV, Wang ZQ, Koh DW, et al. (1998) Poly(ADP-ribose) polymerase null mouse cells synthesize ADP-ribose polymers. J Biol Chem 273: 30069-30072.
40. Ame JC, Rolli V, Schreiber V, Niedergang C, Apiou F, et al. (1999) PARP-2, a novel mammalian DNA damage-dependent poly (ADP-ribose) polymerase. J Biol Chem 274: 17860-17868.
41. Johansson M (1999) A human poly (ADP-ribose) polymerase gene family (ADPRTL): cDNA cloning of two novel polys (ADP-ribose) polymerase homologues. Genomics 57: 442-445.
42. Cantarella L, Gallifuoco A, Malandra A, Martínková L, Spera A, et al. (2011) High-yield continuous production of nicotinic acid via nitrile hydratase-amidase cascade reactions using cascade CSMRs. Enzyme Microb Technol 48: 345-350.
43. Michael C, Lehninger, Albert L, Nelson, David R (2000) Lehninger principles of biochemistry. New York: Worth Publishers.
44. Wan P, Moat S, Anstey A (2011) Pellagra: A review with emphasis on photosensitivity. Br J Dermatol 164: 1188-1200.
45. Ishii N, Nishihara Y (1981) Pellagra among chronic alcoholics: Clinical and pathological study of 20 necropsy cases. J Neurol Neurosurg Psychiatry 44: 209-215.
46. Offermanns S, Schwaninger M (2015) Nutritional or pharmacological activation of HCA (2) ameliorates neuroinflammation. Trends Mol Med 21: 245-255..
47. Chai JT, Digby JE, Choudhury RP (2013) GPR109A and vascular inflammation. Curr Atheroscler Rep 15: 325.
48. Graff EC, Fang H, Wanders D, Judd RL (2016) Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metab Clin Exp 65: 102-113.
49. Wakade C, Chong R (2014) A novel treatment target for Parkinson's disease. J Neurol Sci 347: 34-38.
50. Santolla MF, De Francesco EM, Lappano R, Rosano C, Abonante S, et al. (2014) Niacin activates the G protein estrogen receptor (GPER)-mediated signalling. Cell Signal 26: 1466-1475.
51. Laurence LB, John SL, Keith P (2005) Goodman & Gilman’s The Pharmacological Basis of Therapeutics. New York: McGraw-Hill.
52. Goldie C, Taylor AJ, Nguyen P, McCoy C, Zhao XQ, et al. (2016) Niacin therapy and the risk of new-onset diabetes: A meta-analysis of randomised controlled trials. Heart 102: 198-203.
QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- International Journal of Radiography Imaging & Radiation Therapy (ISSN:2642-0392)
- Archive of Obstetrics Gynecology and Reproductive Medicine (ISSN:2640-2297)
- Chemotherapy Research Journal (ISSN:2642-0236)
- International Journal of Diabetes (ISSN: 2644-3031)
- Journal of Ageing and Restorative Medicine (ISSN:2637-7403)
- Journal of Infectious Diseases and Research (ISSN: 2688-6537)
- Advance Research on Endocrinology and Metabolism (ISSN: 2689-8209)


