Case Report
Preoperative Endocrinological Management of Hypothalamic and Pituitary Tumors in Childhood: A Case Report
3124
Views & Citations2124
Likes & Shares
The main objective of this case report is to review the latest evidence and to highlight the importance of adequate perioperative endocrinological management of hypothalamic-pituitary tumors. A literature review is carried out about a clinical case of a 2-year-old male patient who presented with a voluminous seller tumor with suprasellar extension. He had an unusual clinical presentation with unexplained and frequent falls, ataxia, and functional decline associated with global developmental delay and percentile deflection in the height and weight curves one year prior to diagnosis. Central hypothyroidism was diagnosed preoperatively and treated with intravenous levothyroxine.
After surgical treatment, the patient developed adipose diabetes insipid us with complicated electrolyte management, secondary hypothyroidism, and secondary adrenal insufficiency, which were treated without complications. The pathology report confirmed an Adamantinomatous craniopharyngioma. Treatment in these infrequent pathologies must be individualized and carried out by an experienced multidisciplinary team, given the high morbidity and the sequelae produced by hypothalamic-pituitary injury after tumor exeresis. Surgical treatment continues to be the initial approach, the role of radiotherapy as an initial adjunct therapy is discussed.
Keywords: Craniopharyngioma, Seller tumor, Presurgical hypothyroidism, Panhypopituitarism, Case report
INTRODUCTION
Tumors in the hypothalamic-pituitary region represent 10% of all intracranial tumors in pediatric age; craniopharyngiomas are the most frequent (80-90%) [1]. Tumors can be benign (craniopharyngiomas, pituitary adenomas), malignant (germ cell tumors), or non-neoplastic (pituitary hyperplasia, Rathke's bursa cyst).
The tumors mentioned above are commonly diagnosed by visual or neurological alterations due to mass effect and, less frequently, by associating signs of hypothalamic or pituitary dysfunction [1]. Age of presentation varies between 5-14 years and 50-75 years [2]. Treatment depends on the histological type of lesion. Although the initial approach is usually surgical, it can be exclusively medical (prolactinomas and pituitary hyperplasia) or require only periodic follow-up (small incidentalomas, pars intermedia cysts) [1].
Although most craniopharyngiomas are benign tumors, many are associated with high morbidity rates due to their proximity to vital structures [3]. Age of presentation and debut form stands out in this case.
CLINICAL CASE
A 2-year-old Caucasian male patient without notable perinatal records presents with a one-year history of frequent falls, ataxia, and functional decline. He showed global developmental delay mainly in motor and language skills, with regression of age-appropriate behaviors and deflection in the height and weight curves in the percentile tables in the last year.
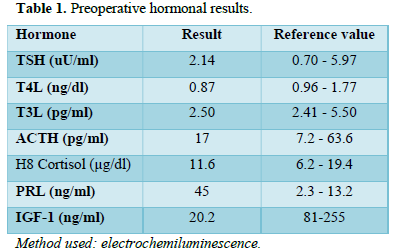
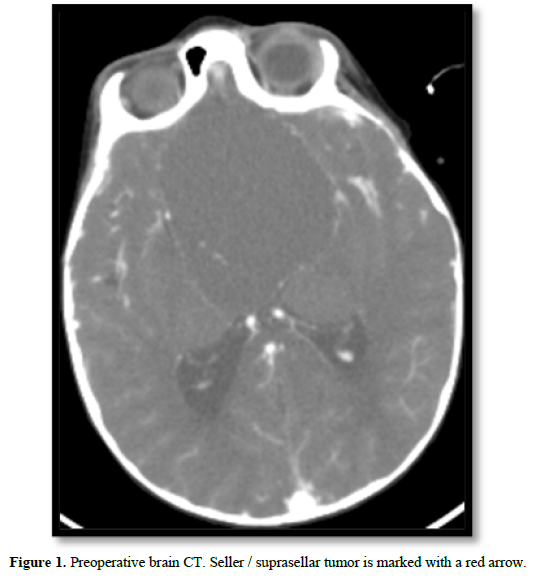
Brain magnetic resonance imaging (MRI) was performed, and a voluminous seller tumor process with suprasellar extension and a cystic component of 75x58x56 mm was observed with a significant mass effect. Figure 1 shows Computed Tomography (CT) images of the brain since we do not count with the first MRI. He was admitted to the Pereira Rossell Hospital Center (CHPR), where an urgent hormonal evaluation was performed (Table 1).
In the preoperative period, intravenous levothyroxine and hydrocortisone 50 mg/m2/day (stress dose) were started due to symptomatic hypoglycemia. However, posterior hour 8 cortisol was found in the gray area, therefore not confirming adrenal insufficiency. Since precise dose conversions have not been established, 80% of the oral dose of levothyroxine (5µg/kg/dose) was administered as suggested by the American Thyroid Association (ATA) guidelines [4]. Transcranial surgery was performed with complete tumor resection 12 h after levothyroxine administration.
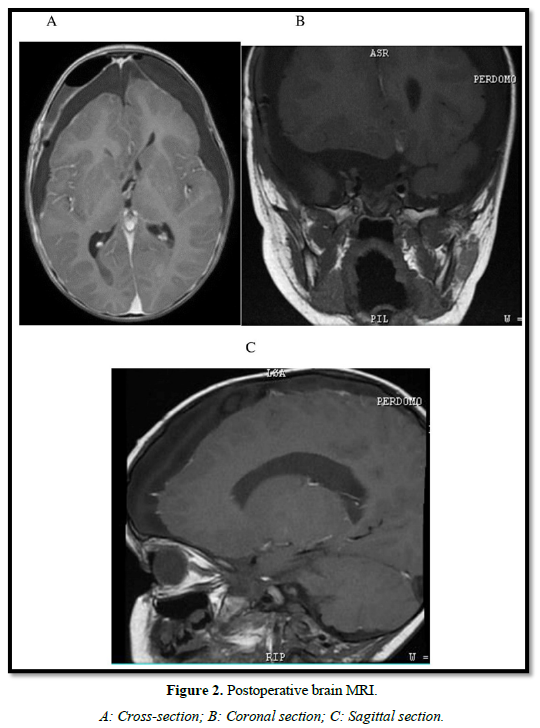
As postsurgical endocrinological complications, he added diabetes insipidus and the maintained panhypopituitarism. Additional postoperative complications were: hypothalamic adipsia, cerebrospinal fluid fistula (CSF), bacterial meningitis, and CSF candidiasis. Postsurgical skull MRI images are shown in Figure 2. The pathology report confirmed an Adamantinomatous Craniopharyngioma.


DISCUSSION
Craniopharyngiomas are rare benign solid or solid-cystic epithelial tumors that develop from the remains of Rathke bursa [1]. Represent 80-90% of the seller/suprasellar area tumors in childhood and 6-13% of all Central Nervous System (CNS) tumors. Have an incidence of 0.13 to 2 cases per 100,000 people / year [1]. Present a bimodal distribution with a peak incidence between 5-15 years and another in the fifth decade of life [5].
As for our case, even if it is not within the most frequent age of presentation, in a study in Hong Kong, China, on CNS tumors in children under three years of age, a craniopharyngioma was observed on a child between 0-1 years old and three cases in children between 2-3 years [6]. Although they are rare, there are case reports of congenital and neonatal craniopharyngiomas.
Usually presents with neurological and structural symptoms, visual impairment due to optic pathway involvement. Even though 80% of patients show several pituitary deficits at the time of diagnosis, that is not the main reason for consultation. Among hormonal insufficiencies, growth hormone deficiency is the most frequent (75% of cases) [3]. Aligned with the case findings presented, in a series of cases where 152 pediatric patients with craniopharyngiomas were studied, 17 patients (11.1%) presented march disturbances (without specifying the type of alteration) [7].
Before surgery, tumors of the seller/suprasellar area require a hormonal evaluation: IGF-1, FSH, LH estradiol, or testosterone (only in pubertal age or if there are signs of precocious puberty), TSH, FT4, ACTH, cortisol hour 8 and PRL.
Surgery is the primary treatment in most craniopharyngiomas, with complete resection being the target without sequelae [2]. Benefits of a surgical approach include anatomopathological diagnosis, relief of structural symptoms, and resection of tumor mass safely, avoiding complications as much as possible [2].
Preoperatively, all patients must receive stress corticosteroid doses, initiated before surgery and in elevated doses during surgery, and continue at progressively lower doses for a time depending on the estimated risk of hypothalamic-pituitary damage. If not administered, surgical pituitary damage can cause an adrenal crisis.
The proposed regimen for corticoid replacement is hydrocortisone 30 mg/m2/intravenous the day before surgery, 100 mg/m2/iv the day of surgery, decreasing progressively on postoperative days until reach oral maintenance dose [1]. It is also necessary to treat the additional hormonal deficits, the thyroid axis the most important one. However, our patient could not be treated given the surgical urgency. In a most extensive retrospective cohort study by Komatsu [8] was evidenced that the dose of levothyroxine varies with age. In the case of secondary hypothyroidism, the required dose is less than that for primary hypothyroidism. It is essential to control natremia and fluid balance in the postoperative period since hydro electrolytic disturbances, especially diabetes insipidus, are the most frequent postoperative complications (50-55 %) [1,9-11].
Additionally, other pituitary hormone deficits must be addressed. Evaluation is recommended six weeks after surgery, while corticosteroid therapy must be maintained. Hydrocortisone is the therapy of choice for children due to its low potency and short half-life that minimize growth impairment among several adverse effects. Secondary adrenal insufficiency replacement dose is 8-10 mg/m2/day orally (somewhat lower than in primary disease), ideally administered every 8 h, adjusting according to clinical response and anthropometry measures [12].
In stressful situations, physiological cortisol secretion increases substantially; therefore, in patients with adrenal insufficiency (primary or secondary), the family must be informed and trained to manage glucocorticoid administration to avoid an adrenal crisis. If fever, vomiting, diarrhea, inadequate oral intake, dental interventions, burns, or mild-moderate trauma, it is necessary to double or triple hydrocortisone dose [11]. Growth hormone treatment is safe, effective, and does not affect tumor relapse and progression rates [10].
Life quality and psychosocial integration are affected by sequelae produced by the tumor and its treatment, including visual impairment, behavioral and cognitive function disorders, and hypothalamic and hormonal secretion disturbances. Patients with craniopharyngioma have 3 to 19 time’s higher cardiovascular mortality compared to the general population [9]. Overall survival rate at 20 years is impaired in patients with hypothalamic involvement. Hypothalamic obesity has a significant negative impact on the quality of life and long-term survival.
An experienced multidisciplinary team is required for the optimal treatment of pediatric patients with craniopharyngiomas [13]. Although the optimal management of craniopharyngiomas remains controversial, first-line treatment usually consists of surgical resection. Complete removal of the tumor provides a high rate of long-term stability; however, aggressive surgery is associated with a significant incidence of complications, as evidenced in our patient.
Radiotherapy (RT) is currently used after limited surgery and achieves excellent long-term tumor control. This evidence may lead to changes in the standard protocol of management in the future [14].
CONCLUSION
Treatment of craniopharyngiomas carries high morbidity due to its proximity to vital structures. The current initial approach is still surgery, being at the same time controversial and individualized to each patient. Given the multiple complications, assessment by an experienced multidisciplinary team is relevant.
- Guerrero-Fernández J (2020) In: Manual of diagnosis and therapy in Pediatric Endocrinology. 1.1. Madrid: ERGON; 2020. pp: 258-282. Available online at: https://ergon.es/wp-content/uploads/2020/06/primeras.pdf
- Harsh AGR, Recht LD, Marcus KJ (2021) Craniopharyngioma. 2021: 1-17.
- García-García E, González-Aguilera B, Gros N, Romero-Lluch A, Jiménez-Varo I, et al. (2014) Diagnóstico y tratamiento endocrinológico de las lesiones del área selar en la edad pediátrica. Endocrinol Nutr 61(7): 359-365.
- Jonklaas J, Bianco AC, Bauer AJ, Burman KD, Cappola AR, et al. (2014) Guidelines for the treatment of hypothyroidism: Prepared by the American thyroid association task force on thyroid hormone replacement. Thyroid 24(12): 1670-1751.
- Venegas E, Concepcion B, Martin T, Soto A (2015) Guía práctica del manejo y tratamiento de los craneofaringiomas y otras lesiones paraselares. Endocrinol Nutr 62(1): e1-e13.
- Liu APY, Shing MMK, Yuen HL, Li CH, Ling SC, et al. (2015) Central nervous system tumors in Chinese children under the age of 3: A population study. J Pediatr Hematol Oncol 37(2): 94-103.
- Ciurea A, Saceleanu V, Mohan A, Moreanu MS, Toader C (2020) Craniopharyngiomas in children-experience of consecutive 152 operated cases. Acta Endocrinol (Copenh) 16(1): 103-109.
- Komatsu R, Karimi N, Zimmerman NM, Sessler DI, Bashour CA, et al. (2018) Biochemically diagnosed hypothyroidism and postoperative complications after cardiac surgery: A retrospective cohort analysis. J Anesth 32(5): 663-672.
- Müller HL, Merchant TE, Puget S, Martinez-Barbera JP (2017) New outlook on the diagnosis, treatment and follow-up of childhood-onset craniopharyngioma. Nat Rev Endocrinol 13(5): 299-312.
- Jensterle M, Jazbinsek S, Bosnjak R, Popovic M, Zaletel LZ, et al. (2019) Advances in the management of craniopharyngioma in children and adults. Radiol Oncol 53(4): 388-396.
- Grau G, Vela A, Rodríguez Estévez A, Rica I (2019) Insuficiencia suprarrenal. Protoc diagn ter pediatr. 1: 205-215.
- Gutiérrez PL (2019) Hipocortisolismo. Sospecha de insuficiencia suprarrenal. En: Julio Guerrero, Isabel González. Manual de diagnóstico y terapéutica en Endocrinología Pediátrica. v 1.1. Madrid: ERGON pp: 693-713.
- Garré ML, Cama A (2007) Craniopharyngioma: modern concepts in pathogenesis and treatment. Curr Opin Pediatr 19: 471.
- Minniti G, Esposito V, Amichetti M, Enrici RM (2009) The role of fractionated radiotherapy and radiosurgery in the management of patients with craniopharyngioma. Neurosurg Rev 32(2): 125-132.
-
 Table 1
Table 1
QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Renal Transplantation Science (ISSN:2640-0847)
- Oncology Clinics and Research (ISSN: 2643-055X)
- International Journal of AIDS (ISSN: 2644-3023)
- Dermatology Clinics and Research (ISSN:2380-5609)
- Journal of Alcoholism Clinical Research
- International Journal of Anaesthesia and Research (ISSN:2641-399X)
- Journal of Cardiology and Diagnostics Research (ISSN:2639-4634)


