2659
Views & Citations1659
Likes & Shares
Adult onset foveo-macular vitelliform dystrophy is a relatively uncommon condition and often misdiagnosed, as the true vitelliform dystrophy is rare in adults. We describe one case of AOFVD; the case underwent a complete ophthalmic examination, fluorescein angiography, systemic evaluation including physical examination and laboratory examination. We did not find any associated systemic disease in our case in spite of extensive investigations.
INTRODUCTION
Adult onset
Foveo-macular vitelliform dystrophy is a condition that presents classically as
bilateral, symmetrical, greyish yellow round or oval lesions within the macular
area best known as ‘poached egg’ appearance [1]. These lesions are mildly
elevated and are one-third to half disc diameter in size. The onset of disease
is usually between 30 and 50 years of age with variable genetic inheritance,
although some have suggested an autosomal dominance inheritance pattern. The
mutations were observed in the BEST1 and PRPH gene respectively. Patients with
AOFVD typically present with symptoms of mild blurred vision or normal vision
and mild metamorphopsia and a mild red-green dyschromatopsia seen in later
stages of the disease [2].
Results of
diagnostic testing show a normal or mildly subnormal electroretinogram (EOG)
[3]. Fluorescein angiography (FA) shows a hypo fluorescent area corresponding
to vitelliform lesion and surrounding ring of hyper fluorescence [4]. Optical
Coherence Tomography (OCT) show the vitelliform lesion as being located in the retinal
pigment epithelium (RPE) layer or between the RPE and photoreceptor layer.
Vitelliform macular dystrophy is a disease of the retinal pigment epithelium
which in later stages known as ‘scrambled egg appearance, may lead to chorio retinal
atrophy [1].
In 1977
Fisherman et al described 3 patients above the age of 45yrs with bilateral
vitelliform lesions with normal EOG findings and visual acuity of 20/100 or
better were termed as ‘pseudo vitelliform macular degeneration’. On fluorescein
angiography found a hyper fluoroscent area around the fovea and was
hypothesized due to leakage from perifoveolar capillaries. The authors thus
emphasised on the use of EOG to differentiate between pseudovitelliform
dystrophy from Best’s disease [5].
Kingham and
Lochen described 6 cases of vitelliform dystrophy with normal EOGs. Angiography
in their cases revealed hyper fluorescent areas due to pigment epithelial
defect thus making the emphasis on leakage from choroid rather than perifoveal
capillary network [6].
Gass studied 9
cases of what he termed ‘peculiar foveolar macular dystrophy’, having
bilateral, symmetrical, raised yellow sub retinal lesions of 1/3rd
disc diameter size with central pigment spot. The onset was primarily in 30 to
50 years of age group, with symptoms of progressive slight blurring of vision
and metamorphopsia. EOG readings were found to be sub-normal with fluorescein
angiography showing hypo fluorescent lesion or a hyper fluorescent ring with
central foveolar hypo fluorescence. The pathology was found in the retinal
pigment epithelium, with some relationship of this entity to family drusen [7].
CASE
A 60 year old female patient presented on
April 2016 with complaints of diminution of vision both eyes for 5-6 years and
was diagnosed as a case of central confluent drusen with presbyopia at a
private hospital and was prescribed with refractive correction but the
condition worsen. Examination of both the eyes showed a Best-corrected visual
acquity of 6/12 in Right eye and 6/9 in the left eye with normal adnexa, cornea
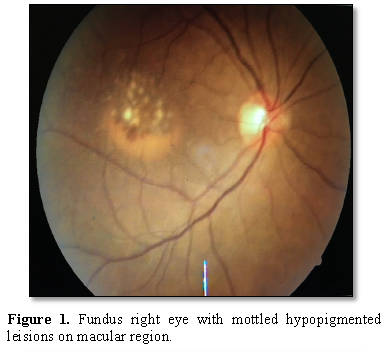
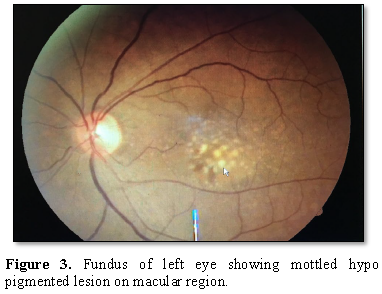
and conjunctiva, anterior chamber and IOP. Fundoscopy revealed a central
confluent drusen with pigmentary dystrophy
at
the macular area.
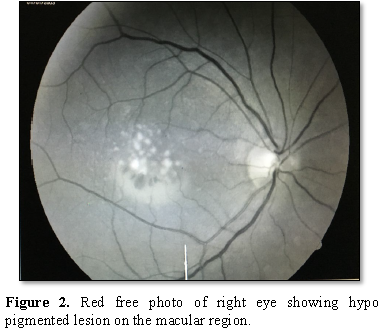
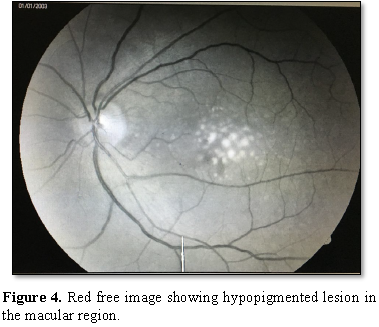
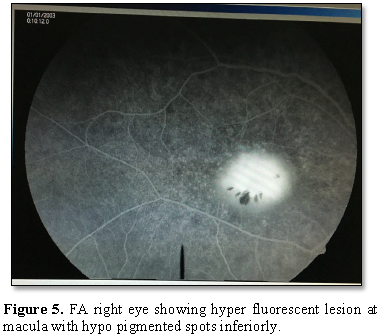
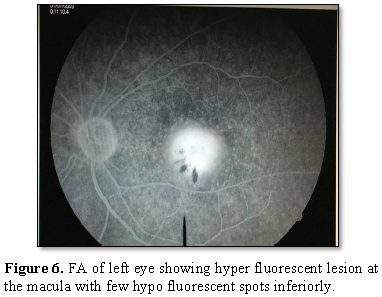
The Red-free
Fluorescein angiography showed a hyper fluorescent
lesion in the macular region approximately the size of one-disc diameter with
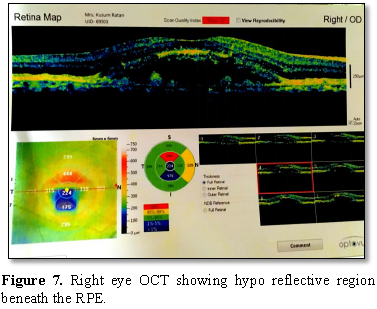
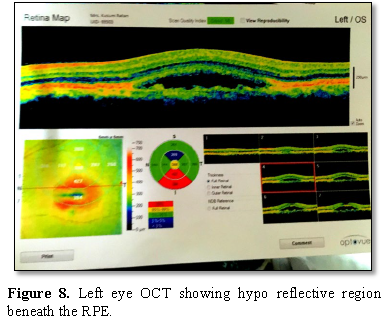
3-4 hypo fluorescent spots present inferiorly. The OCT showed hypo reflective
lesion beneath the RPE in both the eyes at the macular region.
Routine blood investigations showed
deranged lipid profile of increased LDL 0f 114 mg/dl and Kidney function test
with increased uric acid (6.7mg/dl) and alkaline phosphatase (181 U/l).
Electro-oculogram was in normal limits.
She was diagnosed as a case of adult onset
foveo-macular vitelliform dystrophy and was advised to undergo 6-monthly
follow-up for any progression (Figures
1-8).
DISCUSSION
The differential diagnosis of vitelliform
dystrophy is central serous retinopathy, best’s disease, inflammatory retinitis
(toxoplasma retinalis), retinal pigment epithelial detachment, macular drusen
and other macular degenerations.
This disease was an adult onset foveolar
vitelliform dystrophy because:
1. Age
of presentation was 60 years (>30 years).
2. Normal
EOG findings.
3. Small
lesion with the size of one-disc diameter at the macula.
4. Fluorescein
angiography showed hyper fluorescent lesion in the macular region approximately
the size of one-disc diameter.
5. The
visual acuity is minimally subnormal.
The progression of this disease can lead to
a slow decline in visual acuity, with metamorphopsia and red-green dyschromatopsia
in the later stages. The visual acuity decline was found to be symmetrical and
in some cases can be improved with a hyperopic correction, which could be due
to slightly elevated macular lesions. The familial preponderance was also
noted. The fluorescein angiography generally shows a hypo fluorescent fovea
with a hyper fluoroscent ring around the macula. The OCT findings found a
hyoreflective lesion at the level of RPE, showing a neurosensory detachment
above the RPE suggestive of chroidal pathology. The sub retinal exudation is
evidently not associated with a neovascularization or hemorrhages [2]. The
electrical studies are useful in differentiating adult vitelliform dystrophies
from true vitelliform dystrophy as Best’s disease (Table 1).
1.
Best F
(1905) Ueber eine hereditare Maculoaffektion. Beitrage zur Verebunglehre. Z
Augenheilkd 13: 199-212.
2.
Epstein
GA, Rabb MF (1980) Adult vitelliform macular degeneration. Br J Ophthalmol 64:
733-740.
3.
Duetman
AF (1969) Electro-oculography in families with viteliiform dystrophy of the
fovea. Arch Ophthalmol 81: 305-316.
4.
Morse
PH, Maclean AL (1968) Fluorescein fundus studies in hereditary vitelliruptive
macular degeneration. Am J Ophthalmol 66: 485-494.
5.
Fishman
GA, Trimble S, Rabb MF, Fishman M (1977) Pseudo-vitelliform macular
degeneration. Arch Ophthalmol 95: 73-76.
6.
Kingham
JO, Lochen GP (1977) Vitelliform macular degeneration. Am J Ophthalmol 84:
531-536.
7.
Gass
JDM. (1974) A clinicopathological study of a peculiar foveo-macular dystrophy.
Trans Am Ophthalmol Soc 72: 139-156.
-
 Table 1
Table 1

QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Oncology Clinics and Research (ISSN: 2643-055X)
- International Journal of Anaesthesia and Research (ISSN:2641-399X)
- Journal of Spine Diseases
- Journal of Forensic Research and Criminal Investigation (ISSN: 2640-0846)
- Journal of Clinical Trials and Research (ISSN:2637-7373)
- Journal of Alcoholism Clinical Research
- Journal of Renal Transplantation Science (ISSN:2640-0847)


