948
Views & Citations10
Likes & Shares
Molecular
techniques, being fast and precise, have been used in the taxonomic research of
many bacterial genera. The identification of a very heterogeneous group of
species, such as the Burkholderia cepacia
complex (Bcc) and related genera, through basic microbiology techniques, have
been insufficient. In recent years, however, several studies have identified
the biotechnological potential of (Bcc).
Thus, generating techniques such as real-time PCR to be used as a
definitive typing tool has been of utmost importance in research. The main
objective of the present investigation was to identify bacteria producing (Bcc)
antibiotics molecularly, through the analysis of the RecA region by means of
real-time PCR. To fulfil with the objective, DNA extraction and purification
methods were analyzed statistically: High Pure Template Preparation Kit-ROCHE,
boiling and PCI, by means of ANOVA and Tukey, determining a significant
difference (p˂0.001); The ROCHE kit methodology presented the best DNA quality
and concentration results. The boiling method is shown here, as a
low-costalternative to show DNA with
acceptable characteristics and quality for the real-time PCR technique. For
molecular identification, amplification of the 16S regions rRNA-Bcc, Burkholderia sp. (RecA) and B. cepacia-genomovar I (RecA) using
specific primers took place. The analysis of the amplification curves obtained
by real-time PCR confirmed that BC1 belongs to the species Burkholderia cepacia (genomovar I); afterwards, it was confirmed
that BC2 belongs to the genus Burkholderia,
but the genomovar of the organism was not corroborated; however it is asserted
that the strain belongs to some genomovar of Bcc based on the amplification of
the 16S region for Bcc and its biochemical identification.
Keywords: Burkholderia
cepacia complex; RecA gene, genomovar; molecular detection; Real-time PCR
Abbreviations: Bcc: Burkholderia
cepacia Complex; PCI: Phenol-chloroform-isoamyl Alcohol; PCR: Polymerase
Chain Reaction; P16S: 16S rRNA-Bcc Regions; PSPB: Burkholderia sp. (RecA); PBC1: B.
cepacia-genomovar I (RecA); RFLP: Restriction fragment length polymorphism
INTRODUCTION
Burkholderia cepacia is a gram
negative non-fermenting bacillus (BGNNF) identified as a phytopathogen and
currently recognized as an important opportunistic pathogen. It comprises a
very heterogeneous group of phenotypically similar but phylogenetically
distinct species (genomovars) until now a total of 20 genomovars have been
described that makeup what is called the Burkholderia
cepacia complex(Bcc). The basic microbiology techniques have been
insufficient to generate an accurate diagnosis in the identification of
genomovars of Bcc and related genera, taking the PCR technique as a definitive
typing tool [1,2].
The molecular identification techniques in bacteria have
led to the search for candidate genes (5S, 16S, 23S rRNA) and their intergenic
spaces to be used in the taxonomic research of many bacterial genera. However,
the analysis of 16S rRNA to establish phylogenetic differences within the Bcc
is limited. An alternativeis the amplification of the RecA gene fragments,
which presents sufficient nucleotide variation to allow such discrimination
between their variants [3,4].
In recent years, genomovars belonging to the Bcc have
been of great importance in studies relating to agriculture, as biological
control agents with antifungal activity, improved crop yields, production of
antibiotics, bioremediation of landfills, contaminated soils and groundwater;
becoming organisms with a high biotechnological potential [1].
The present investigation identifies molecularly Bcc
bacteria [Burkholderia cepacia
(genomovar I)] through the analysis of the RecA-specific region by real-time
PCR; isolated bacteria, characterized morphologically and biochemically from
soils of the natural regions of Ecuador, capable of producing antibiotics [5].
MATERIALS AND METHODS
Selection of the sample
The present research study was carried out in the Life
Sciences Laboratories of the Universidad Politécnica Salesiana Quito, Sede El
Girón. Two bacterial strains capable of producing antibiotics were selected
from a total of 27 isolated strains, from soils of the Insular-Galápagos region
(Puerto Ayora) and the Sierra-Norte region (Quito-Pichincha) [5].
Analysis of bacterial
concentration
Samples BC1 and BC2 were inoculated in TSB medium at
pH 7 ± 0.2 for 24 hours at a temperature of 30 ± 2 ° C [5]. Using the McFarland
turbidity standards the bacterial concentration of 9 x 108 CFU / mL was
calculated.
Extraction of DNA
from samples
Using three different extraction methods, genomic DNA
was isolated from a total of 60 samples, 10 replicates for each strain and for
each methodology.
DNA extraction and
purification technique with "High Pure Template Preparation Kit" kit
- ROCHE
200 μL of the sample material with 5 μL of lysozyme, was
placed in a 1.5 mL Eppendorf tube free of nucleases; mixed and incubated at 37
° C for 5 minutes to continue with the manufacturer's instructions.
The sample was transferred to a purification tube with
its specific filter and centrifugation was performed for one minute at 8000 x
g. Finally, 200 μL of pre-heated elution buffer at 70° C was added to the
filter tube, centrifuged and stored at -20 ° C for subsequent analyzes [6].
DNA extraction
technique by modified boiling
200µL of the sample material was
placed in a 1.5mL Eppendorf tube free of nucleases; the sample was centrifuged
at 10000 x g for 5 minutes and the supernatant was discarded, the sediment was
suspended in 200 μL of sterile solution of sodium chloride 0.85%, the mixture
was centrifuged at 10000 x g for 5 minutes and the supernatant was discarded.
The sediment was suspended in 200 μL of 1X TE Buffer
and incubated at 95 ° C for 25 minutes. It was then centrifuged at 10000 x g
for 3 minutes and 200 μL of the supernatant containing the DNA was transferred
[7].
DNA extraction
technique with organic solvents Phenol / Chloroform /Modified Isoamyl alcohol
200µL of the sample material was placed in a 1.5mL
Eppendorf tube free of nucleases, the sample was then centrifuged for 2 minutes
at 10000 x g. the supernatant was discarded.
The obtained pellet was then mixed with
576 μL of TE 1X buffer, 30 μL of 10% SDS and 3 μL of Proteinase K (20
mg/mL), 100 μL of 5M Sodium Chloride and 80 μL of CTAB / NaCl. This was then
incubated for 10 minutes at 65° C.
Subsequently, 710 μL of chloroform/isoamyl alcohol
(24:1) was added, mixed and centrifuged for 5 minutes at 10000 x g. The aqueous
phase was transferred to a new microcentrifuge tube and an equal volume of
phenol/chloroform/isoamyl alcohol (25: 24: 1) was placed, centrifuged for 5
minutes at 10000 x g.
The supernatant was transferred to a new
microcentrifuge tube and 0.6 volumes of absolute ethanol were added, the
microcentrifuge tube was vortexed and the supernatant was removed. This was
then followed by placing 400 μL of 70%
ethanol, after a mild vortexing , it was centrifuged for 5 minutes at 10000 xg,
the supernatant was carefully discarded and the pellet was allowed to dry
briefly at 65 ° C, the dry contents of the tube were dissolved in 200 μL of TE
1X Buffer [8].
Quantification and
purity of DNA
The DNA concentration was quantified in a Qubit 2.0
Fluorometer® kit, using the kits: Qubit dsDNA HS Assay Kit (0.2-100 ng) and
Qubit dsDNA BR Assay Kit (2-1000 ng) [9]. The purity of the sample was examined
by the absorbance ratio at 260 nm and 280 nm on a NanoDrop computer [10].
The values of DNA concentration (ng/mL) obtained
were subjected to a logarithmic transformation of base 10. Logarithmic
transformations are usually necessary and appropriate to analyze variables
related to the growth of organisms [11].
Real-time PCR
technique
The DNA extracted from the sample BC1 and BC2 was
amplified with a reaction volume for capillaries with a capacity of 20 μL; each
reaction was carried out with 5 μL per sample and controls, 10.2 μL of Molecular Biology grade water, 0.4 μL of the
First FW, 0.4 μL of the First RW and 4 μL of the Master Mix SYBR Green I [12].
Two controls were used in the amplification run: a negative control PCR grade
water and a positive control Burkholderia
cepacia ATCC®25608.
The 16S rRNA Bcc (P16S) primers used were, F: 5'-
GACTCCTACGGGAGGCAGCAG-3'and R: 5'- CTGATCCGCGATTACTAGCGATTC -3' [13].
Burkholderia sp. (RecA)
(PSP), F: 5'- GTCGGGTAAAACCACGCTG -3' and R: 5'- TCCGCAGCCGCACCTTCA -3' [14]. B. cepacia-genomovar I (RecA) (PBC1), F:
5'- CAGGTCGTCTCCACGGGT -3' and R: 5'- CACGCCGATCTTCATACGA -3' [13].
The real-time PCR reactions were carried out in the
LightCycler 2.0 device, according to the protocol of LC FS DNA MasterPLUS
HY-Pb, 96 reactions. LightCycler (Roche Diagnostics), consisting of 35 cycles
composed of four steps: Denaturation: 95 ° C, 10 min; Alignment: 62 ° C, 10
sec; Extension: 72 ° C, 7 sec; Cooling: 40 ° C, 30 sec [12].
STATISTIC ANALYSIS
DNA concentrations (ng/mL) were compared using
the parametric statistical test DCA - ANOVA together with a Tukey analysis. The
results were processed with the help of the statistical package InfoStat.
Values of p less than 0.01 were
considered significant.
RESULTS AND
DISCUSSION
Quantification and purity of DNA
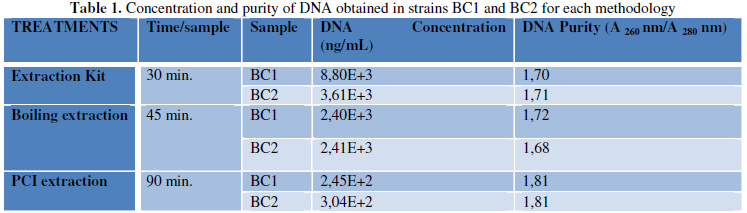
The total concentration and purity of the DNA for each
method are described in Table 1. The
three extraction methods resulted in a good amount of DNA. Bacterial DNA is worked with a sample of up
to 1 to 10 ng, taking into account that the excess in the concentration of DNA
mould in the PCR can lead to non-specific amplifications or failure to amplify
[15]. A greater quantity of DNA was obtained by the Roche commercial method
with an average of 6.205E+3 ng/mL, followed by the boiling method with 2.405E+3
ng/mL and in lesser quantity with the PCI method with 274.5 ng/mL.
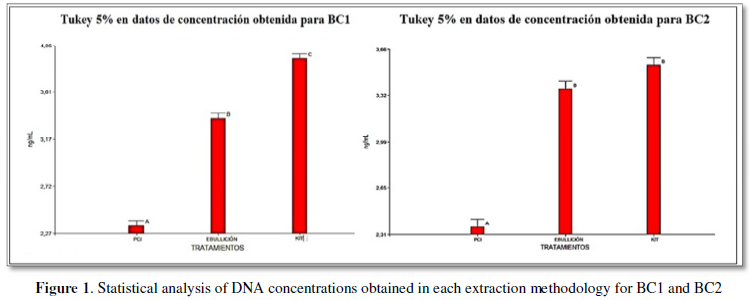
The analysis of variance together with the Tukey test,
evaluated the best DNA extraction technique for each sample as shown in Figure 1, the commercial extraction method
ROCHE tested in strains BC1 and BC2 showed a significant difference compared to
the means of the boiling and PCI methods.
The purity range of the DNA (A260/A280) was between
1.81 and 1.68. For strain BC1, the three methodologies allowed the obtention of
good quality DNA; for strain BC2, the commercial methodology ROCHE and PCI
allowed to also obtain good quality DNA, however, it is observed that in the
boiling technique there is a slight contamination with proteins. The use of
traditional methods has advantages in terms of low costs of reagents and materials, as well as
obtaining a DNA with high performance, however, sometimes the material obtained
is fragmented because the methods can be susceptible to contamination,
variations and errors due to the multiple handling steps [16].
In order to carry out new molecular techniques such as
real-time PCR, the obtention and usage of a high-quality DNA template sequence
(integral and pure) is essential. Therefore, making the correct selection and
application of an appropriate methodology is an integral part of DNA extraction
[17-18].
Molecular
identification of Burkholderia cepacia
by real-time PCR
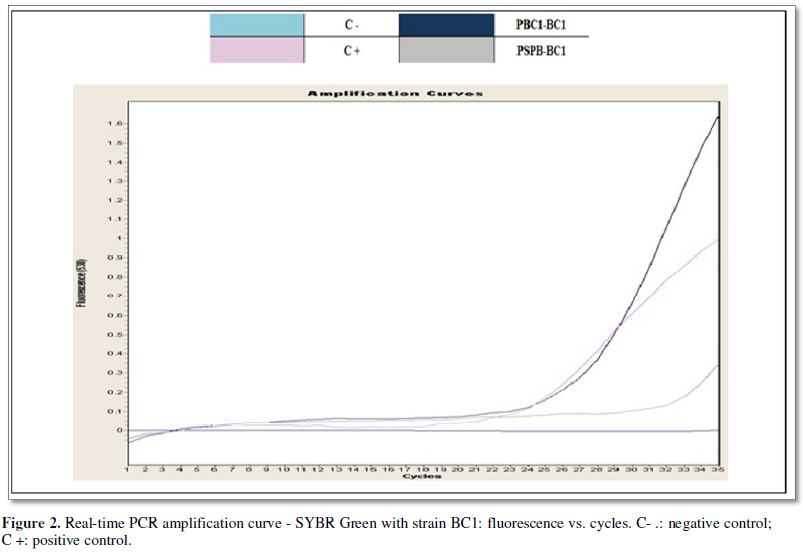
Figure 2. shows the
amplification products for the strain BC1, strain identified biochemically as Burkholderia cepacia with an 88.79%
probability. The amplification showed hybridization with the first primer PSP,
specific for Burkholderia sp.,
corroborating the genus of the species and with the first primer PBC1, specific
for Burkholderia cepacia (Genomovar
I), confirming the species.
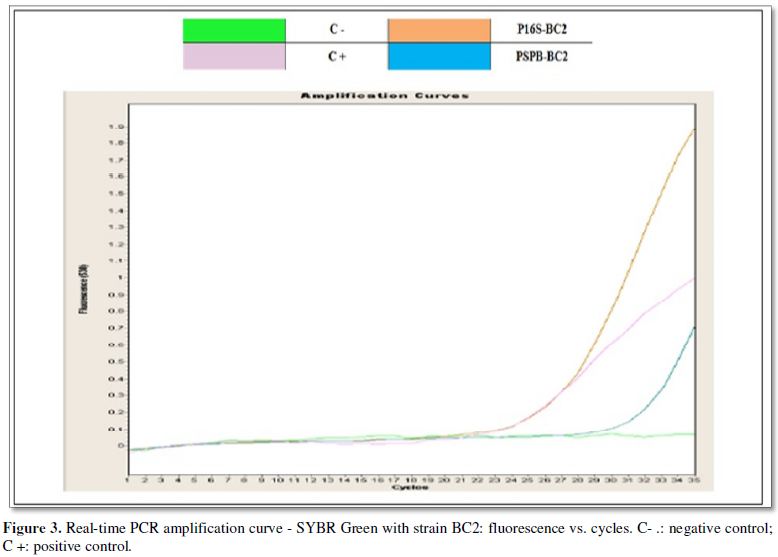
In Figure 3.We
see the amplification products for the strain BC2, strain identified
biochemically as Burkholderia cepacia
with a 99.51% probability. The amplification did not show hybridization with
the first specific primer PBC1 for Burkholderia
Cepacia (Genomovar I), however, the amplification with the first primers
PSPB and P16S, based on these results, was corroborated by the technique of PCR
in real time that the strain BC2 It belongs to the genus Burkholderia, however, it is not identified as the subspecies Burkholderia cepacia (genomovar I), so
it is believed that this strain could be located at a different genomovar
belonging to the heterogeneous group of Bcc.
The identification of B. cepacia is a complex task. This complexity is due to the close
phylogenetic relationship of B. cepacia
with other genera of BGNNF since it is not a single species, but a
heterogeneous group [1,2] . In the study carried out by Canale [3], he argues
that commercial systems of phenotypic identification have significant
variations in their ability to accurately identify Bcc since they can not
differentiate the different genomovars, which is why several studies have made
erroneous identifications of B. cepacia.
In their study, only 1 of 23 phenotypically analyzed samples tested positive
for B. cepacia (4%), by molecular
analysis (PCR-RFLP) 11 of the 23 samples gave positive results for B. cepacia (48%).
Araque et al. [1] concludes that the use of
conventional biochemical methods, complementary biochemical tests, and
non-commercial systems do not allow for correct and clear species-level
discrimination within the Bcc, even though the analysis of the methods showed
moderate sensitivity and specificity ranges compared to other studies
previously reported. However, the use of these methods allow a good
identification of B. cepacia between
isolates of BGNNF, it is also mentioned that to achieve a correct identification
of the strains it is necessary to accompany the phenotypic methodology with
molecular techniques such as real-time PCR (PCR-RFLP), technique that with the
use of specific primers allows a high discriminatory range. Therefore the
relationship between the biochemical methods (conventional tests and galleries)
with the results of PCR, demonstrates the high level of resolution of this
methodology.
The study carried out by Dalmastri et al. [19]
concludes that the RecA species-specific PCR technique for the identification
of Bcc environmental isolates, as B.cenocepacia,
may lead to an underestimation of the organisms belonging to the complex. The
use of isolates from different microbial populations, different geographical locations and sampling
time can present differences in the sensitivity of the method, causing a bias
in the analysis of the populations. However, it explains that this decrease in
sensitivity is not sufficient to establish a deficiency of the method since it
can depend also on the variability of the RecA sequence. In addition, it
mentions that the application of the RecA species-specific technique associated
with different patterns of the RFLP technique increases the sensitivity of the
method. Therefore it considers the analysis RecA-RFLP useful, fast and precise
in the identification of the complex in clinical isolates. Therefore, the
evaluation and optimization of new identification tests for Bcc species should
be performed not only in clinical isolates but also in environmental isolates
in order to improve the detection of these strains in natural habitats.
CONCLUSIONS AND
RECOMMENDATIONS
The comparison of three methods of extraction
of bacterial genomic DNA concluded that the best results were achieved
through the kit protocols and boiling. In this case, boiling is presented as a
viable alternative to the more expensive commercial protocols: besides presenting DNA of good quality (pure
DNA) and adequate concentration values, the extraction time is relatively
short. Current molecular biology techniques such as real-time PCR do not demand
large amounts of DNA because it is a sensitive methodology, but it does demand
integrity and purity [20].
The molecular technique of PCR in real time, through
the analysis of the amplification curves of the region RecA-specific in Burkholderia cepacia, confirmed that
strain BC1 belongs to the species Burkholderia
cepacia (Genomovar I). In turn, it was confirmed that the BC2 strain
belongs to the genus Burkholderia,
but not corroborated the genomovar of the organism. However, it was asserted
that the strain belongs to the group of the complex Burkholderia cepacia based on the amplification of the 16s region
for Bcc and in its biochemical identification.
It was determined that the real-time PCR technique,
through the analysis of the region, allows the molecular identification of
bacterial strains, confirming with precise data biochemical tests that do not
allow a clear discrimination.
It is recommended to evaluate and standardize the
technique of DNA extraction by boiling in Gram-negative bacteria. In this case,
the results obtained from DNA quality in bacteria of the genus Burkholderia were optimal and suitable for
molecular analyses as real-time PCR, in addition, it is presented as a viable
alternative against high-cost commercial methods.
It is necessary to evaluate the strains identified as Burkholderia cepacia (Genomovar I) and Burkholderia sp., belonging to the Bcc,
by using the polymorphism technique in the length of the restriction fragments
(RFLP) of the RecA gene. This is a fast and precise technique, which increases
the sensitivity of the species-specific PCR RecA, in order to identify and
corroborate with the results obtained, and in turn, improve the detection of
isolated strains in natural habitats.
Finally, the antibiotics produced by the strains
belonging to the Burkholderia cepacia
complex must be analyzed and chemically identified, since these bacteria have
proven to possess a high biotechnological potential.
ACKNOWLEDGEMENTS
To Daniel Acurio for his valuable contribution to the data analysis.
1.
Araque Y, Albarado L, Centeno S, Rodríguez L, Vitelli J
(2007) Actividad antibiótica y antifúngica de B. cepacia provenientes de ambientes nosocomiales, Servicio
Autónomo Hospital Universitario "Antonio Patricio Alcalá", Cumaná,
Venezuela. Scielo 35.
2.
Mirambell A (2015) Aspectos microbiológicos de Burkholderia cepacia Complex en
pacientes con fibrosis quística, Tesis publicada como requisito para el grado de
Doctor en Microbiología, Universitat Autònoma de Barcelona, Barcelona,
España.
3. Canale J (2004)
Detección e identificación molecular de genomovares del Complejo Burkholderia cepacia en pacientes con
fibrosis quística del noreste de México, Tesis publicada como requisito para
obtener el grado de Maestro en Ciencias con Especialidad en Biología Molecular
e IngenieríaGenética, Universidad Autónoma de Nuevo León, Nueva León, México.
4.
Fernández A, García C, Sáez J, Valdezate S (2011) Métodos
de identificación bacteriana en el laboratorio de microbiología, Enfermedades Infecciosas y Microbiología
Clínica.
5.
Egas C, Tinajero M (2016) Aislamiento de microorganismos
capaces de producir antibióticos, a partir de suelos de las regiones naturales
del Ecuador,Tesis publicada como requisito para el grado de Ingenieras en
Biotecnología de los Recursos Naturales, Universidad Politécnica Salesiana,
Quito, Ecuador.
6.
Roche
Diagnostics (2012) High Pure Template
Preparation Kit. Rapidly purify genomic DNA for diverse applications.
7.
Mena C (2010) Evaluación de la prevalencia de Listeria monocytogenes en productos
lácteos y embutidos en tres mercados de la ciudad de Quito mediante la técnica
de la reacción en cadena de la polimerasa en tiempo real, Tesis publicada como
requisito para el grado de Licenciatura en Microbiología Clínica y Aplicada, Pontificia Universidad Católica del Ecuador,
Quito, Ecuador.
8.
Ausubel F, Brent R, Kingston R, Moore D, Seidman J, et
al. (2003) Current Protocols in
Molecular Biology: Preparation and Analysis of DNA, Wiley, J & Sons,
New York, USA.
9.
Thermo Fisher (2015) Qubit Assays.
10.
Chacón R (2016) Extracción, Purificación y Cuantificación
de Ácidos Nucléicos.Presentación del 8vo Congreso a Distancia: Técnicas de
Biología Molecular, Nov.Dec, Perú.
11. Sánchez-Otero J
(2013) Introducción a la Estadística en las Ciencias Biológicas: Transformación
de Datos, Quito, Ecuador.
12. Chiluisa-Utreras
V, Echeverría A (2017) Identificación y cuantificación de Salmonella sp. y ADNr 16S bacteriano mediante PCR en tiempo real en
muestras de alimentos. BionaturaVol 2, Num 1: 241-244.
13.
Mahenthiralingam
E, Bischof J, Byrne S, Radomski C, Davies J, et al. (2000) DNA-Based Diagnostic
Approaches for Identification of Burkholderia
cepacia Complex, Burkholderia vietnamiensis, Burkholderia multivorans,
Burkholderia stabilis and Burkholderia cepacia Genomovars I and II. J Clin Microbiol 9: 3165-3173.
14. Henry D, Mahenthiralingam E,
Vandamme P, Coenye T, Speert D (2001) Phenotypic Methods for Determinig
Genomovar Status of the Burkholderia
cepacia Complex. J Clin Microbiol
39: 1073-1078.
15.
Tamay de Dios L, Ibarra C, Velasquillo C (2013)
Fundamentos de la Reacción en Cadena de la Polimerasa (PCR) y de la PCR en
Tiempo Real, Investigación en Discapacidad 2: 70-78.
16.
Cornejo
A, Serrato A, Rendón B, Rocha M (2014) Herramientas
Moleculares Aplicadas en Ecología: Aspectos Teóricos y Prácticos, México D.F,
México.
17.
Fraga J, Rodríguez J, Fuentes
O, Castex M, Fernández A (2004) Comparación entre 5 métodos para la extraccion
de ADN de Triatomineos: su utilización en la técnica de ADN polimórfico
amplificado al azar (RAPD). Scielo 56:
208-213.
18.
Balmes G, Serrano A, Arís A (2009) Evaluación de cuatro
métodos de extracción de DNA bacteriano de muestras de contenido ruminal. AIDA: 778-780.
19.
Dalmastri C, Pirone L, Tabacchioni S, Bevivino A,
Chiarini L (2005) Efficacy
of species-especific recA PCR test in the identification of Burkholderia cepacia Complex
enviromental isolates. FEMS
Microbiology Letters 246: 39-45.
20. Chiluisa-Utreras
V, Cabrera M, Valladares P (2017) Detección de Listeria spp. yListeria monocytogenes en muestras de
leche cruda y quesos artesanales respectivamente, mediante PCR en Tiempo Real. Respuestas 22: 67-75.
-
 Table 1
Table 1