3419
Views & Citations2419
Likes & Shares
In the current study, a green approach to the synthesis and functionalization of GO-ECH-DETA hybrids Nano composite by the reaction of graphite oxide (GO) with epichlorohydrin (ECH) as a coupling agent and diethylenetriamineas (DETA) a ligand, were prepared and employed for endotoxin adsorption and removal from aqueous solution. This kind of Nano composite undoubtedly shows promising applications in the fabrication of multi-functional materials and can be used as a potential candidate for removal of PLS from aqueous solution. Multilayered porous hierarchical structure of GO-ECH-DETA hybrids was prepared via in situ hydrothermal growth of epichlorohydrin (ECH) as a coupling agent and diethylenetriamineas (DETA) a ligand Nano polymer within interlayer space of reduced graphene oxide, which demonstrated a high specific area of 138 m2 g-1 and rational porous structures. Batch adsorption studies showed that the product possesses superior adsorption capacity of endotoxin from aqueous solution. This is a simple and cheap procedure, has proven to remove endotoxins without affecting any significant losses in protein yields and biological activities. The physicochemical properties of the Nano composite were fully characterized, adsorption equilibrium and kinetic analysis indicated that the adsorption isotherm was well fitted by Langmuir isothermal model with the maximum adsorption capacity of 149.45 m2 g-1, the kinetics of the endotoxin adsorption process was shown to follow the pseudo-second-order model. The results showed that the optimum condition for endotoxin removal was obtained at pH of 5.52, GO-ECH-DETA dosage of 100 mg L-1, contact time of 60 min and endotoxin concentration of 150.0 endotoxin units per milliliter (EU mL-1).
Keywords: Graphene oxide, Epichlorohydrin, Diethylenetriamineas, Nanocomposites, Endotoxin, Adsorption
INTRODUCTION
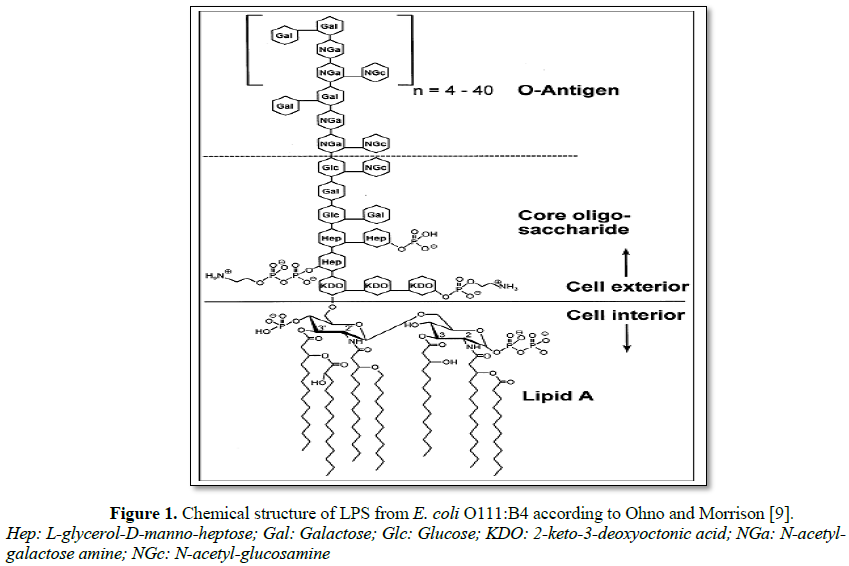
In the biotechnology and pharmaceutical industries, recombinant DNA products such as peptides and proteins are produced by the Gram-negative bacteria such as Escherichia coli. These products are usually contaminated with endotoxins [10]. For this reason, these proteins must be purified from endotoxin, to prevent any side effects induced when administered to humans. However, LPS are very stable molecules, resisting to extreme temperatures and pH values in comparison to proteins [11-13]. Many different procedures have been used for the accumulation and removal of LPS from proteins. These include two-phase extractions, affinity resins, ultrafiltration, membrane adsorbers, hydrophobic interaction chromatography, and ion exchange chromatography. These techniques provide different efficiency degrees of LPS separation from proteins, is highly dependent on the properties of the protein of interest [14]. LPS structure composed of a hydrophilic polysaccharide moiety, which is covalently linked to a hydrophobic lipid moiety (Lipid A) (Figure 1) [10]. Endotoxins from most species are composed of three distinct regions: the O-antigen region, a core oligosaccharide and Lipid A (LipA) as depicted in Figure 1.
The most biological activate part of endotoxin is lipid A, and indeed is responsible for its toxicity. Endotoxin is composed of b-1,6-linked D-glucosamine residues, covalently linked to 3-hydroxy-acyl substituents with 12-16 carbon atoms via amide and ester bonds. These further esterified with saturated fatty acids. This hydrophobic part of endotoxin adopts an ordered hexagonal arrangement, resulting in a more rigid structure compared to the rest of the molecule [13,14]. The core oligosaccharide has an inner 3 deoxy-D-manno-2-octulosonic acid (KDO) - heptose structure region and an outer hexose region. In E. coli species, five different core types are known and Salmonella species share only one core structure. The core region close to lipid A and lipid A itself are partially phosphorylated (pK1=1.3, pK2=8.2 of phosphate groups at lipid A). Thus endotoxin molecules exhibit a net negative charge in common protein solutions [12]. The O-antigen is generally composed of a sequence of identical oligosaccharides (with three to eight monosaccharides each), which are strain specific and determinative for the serological identity of the respective bacterium [8]. The endotoxin monomer molar mass range from 10 to 20 kDa, owing to the variability of the oligosaccharide chain; even extreme masses of 2.5 (O-antigen-deficient) and 70 (very long O-antigen) kDa can be found. There are various forms of endotoxins supra-molecular aggregates in aqueous solutions due to their amphipathic structures. Based on molecular dynamics, the three-dimensional structure of endotoxin, especially the long surface antigen, is much more flexible than the globular structure of proteins [13]. These aggregates result from non-polar interactions between lipid chains as well as of bridges generated among phosphate groups by divalent cations [1]. The aggregate structures have been studied by different techniques such as electron microscopy, X-ray diffraction, FT-IR spectroscopy and NMR. Results from these studies have shown that, in aqueous solutions, endotoxins can self assemble in a variety of shapes, such as lamella, cubic, and hexagonal inverted arrangements, with diameters up to 0.1 mm and 1000 kDa and high stability depending on the solution characteristics (pH, ions, surfactants, etc) [1,2].
The commonly approved techniques used by FDA for endotoxin detection are the rabbit pyrogen test and Limulus Amoebocyte Lysate (LAL) assay [15,16]. The rabbit pyrogen test, is an old technique used in 1920s, involves measuring the rise in temperature of rabbits after intravenous injection of a test solution. Due to its high cost and long turnaround time, the use of the rabbit pyrogen test has diminished and is now only applied in combination with the LAL test to analyze biological compounds in the earlier development phase of parenteral devices. Today the most popular endotoxin detection systems are based on LAL, which is derived from the blood of horseshoe crab, Limulus polyphemus and clots upon exposure to endotoxin. The simplest form of LAL assay is the LAL gel-clot assay. When LAL assay is combined with a dilution of the sample containing endotoxin, a gel will be formed proportionally to the endotoxin sensitivity of the given assay. The endotoxin concentration is approximated by continuing to use an assay of less sensitivity until a negative reaction (no observable clot) is obtained. This procedure can require several hours [16]. The concentration of 0.5 EU/mL was defined as the threshold between pyrogenic and non-pyrogenic samples [16].
In addition to the gel-clot technique, scientists have also developed two other techniques: turbidimetric LAL technique and the chromogenic LAL technique. These newer techniques are kinetic based, which means they can provide the concentration of endotoxin by extracting the real-time responses of the LAL assay. Turbidimetric LAL assay contains enough coagulogen to form turbidity when cleaved by the clotting enzyme, but not enough to form a clot [17]. The LAL turbidimetric assay, when compared to the LAL gel-clot assay, gives a more quantitative measurement of endotoxin over a range of concentrations (0.01 EU/mL to 100.0 EU/mL). This assay is based on the turbidity increase due to protein coagulation related to endotoxin concentration in the sample. The optical densities of various test-sample dilutions are measured and correlated to endotoxin concentration helped by a standard curve obtained from samples with known amounts of endotoxin [18]. A kinetic chromogenic substrate assay differs from gel-clot and turbidimetric reactions because the coagulogen is partially or completely replaced by a chromogenic substrate [19]. When hydrolyzed by the pre-clotting enzyme, the chromogenic substrate releases a yellow-colored substance known as p-nitroaniline. The time required to attain the yellow substance is related to the endotoxin concentration [18]. However, kinetic turbidimetric and chromogenic tests, although more accurate and faster than the gel-clot, can not be used for fluids with inherent turbidity such as blood and yellow-tinted liquids, e.g. urine and their performance may be compromised by any precipitation from solution [19]. Therefore, different new methods for detection of endotoxin in different samples have been studied and approved [20,21].
Carbonaceous nanomaterials, including carbon nanotubes (CNTs) and graphene based materials (GBMs) such as graphene oxide (GO), hold significant promise in engineering and medicine due to their intrinsic electro-mechanical properties. Graphene and graphine oxide are the most basic form of carbon, it is composed of sp2 bonded carbon atoms arranged in a hexagonal arrangement in a 2D plane [22]. The lattice of graphene consists of two interleaved triangular shaped carbon sublattices. The sublattices overlap in such a way that carbon atom from one sublattice is at the centroid of the other sublattice. Graphene has been utilized in many engineering and industrial applications and graphene-based polymer nanocomposites exhibit superior promising properties. For example, graphene-based polymer composites show better thermal, mechanical and electrical properties than the normal polymer [23,24]. It has been shown that the mechanical and electrical properties of graphene-based polymer composites are much better in comparison to clay or other carbon filler-based polymer composites [25-29]. One of the main applications of graphene sheets is use as reinforcement agents for the preparation of nanocomposites with different types of polymers. Other than mechanical properties, electrical and thermal properties of the polymeric matrix can also be enhanced. It is a fact that the graphene-based nanocomposites present improved properties compared to the original raw form of graphene. Graphene nanocomposites with polysaccharides such as chitosan have many diverse new applications. Polysaccharide exist both as linear or branched polymers, since their repeating monosaccharide units are connected via O-glycosidic bonds [30]. Their properties, including gelation, water solubility and other surface properties depend on the type of monosaccharide composition. Advantages such as abundance in nature, biocompatibility, biodegradability, easy functionalization and relatively easy isolation from their natural sources have led to their study and use in several applications, especially in the field of drug delivery and biomaterials [31]. Another application of graphene derivatives with polysaccharide is the use in accumulation and removal of various types of pollutants from wastewater effluents [32]. Graphene nanocomposites with chitosan have been used for the removal of dyes [33,34], heavy metal ions [35] and pharmaceutical compounds [36] from aqueous solutions. Despite the intriguing properties of polysaccharides, their poor mechanical properties limit their applications. Nanofillers such as graphene are known to improve the properties of raw polymers, not only the mechanical but also the thermal and electrical properties [37,38]. Moreover, the effects of the incorporation of graphene and graphene oxide together in raw polymers have been extensively studied; mostly synthetic polymers reinforced by graphene and graphene oxide find several improvements in properties such as mechanical strength, thermal stability, gas barrier properties, electrical and thermal conductivity, etc. [39-46]. Cationic polymers which are useful as flocculants are prepared by condensation reaction of a dialkylamine, dicyandiamide and a polyalkylenepolyamine, with a difunctional epoxide. Functionalization of graphene oxide (GO) by crown ether moiety to attach Li⁺ in the cage of five oxygens is a useful tool to achieve 2D material for Li ion battery. The attachment of crown ether occurs only through the reaction with epoxy groups of GO. Epichlorohydrin is used to increase the number of the epoxy group to enhance and precise control of the Li⁺ content for tuning the activation energy of Li⁺ migration [47].
The final properties of nanocomposites depend on various factors; the most important is the interfacial bonding between the filler and the matrix. Poor adhesion can lead to aggregates of the nanofillers or gaps between the surface of the composites components, acting as stress concentration points and therefore causing premature failure of the materials. Besides, the compatibility between nanofiller and matrix, the geometrical and the aspect ratio of the fillers play a similarly important role. Graphene possesses a high surface area, high aspect ratio and high strength which are reasons for the enhanced performance of its nanocomposites. Large graphene or GO flakes with high surface areas have proved to be more efficient reinforcing agents than similar structures with smaller aspect ratio.
Some commonly used techniques have been extensively used for removing endotoxin contaminants are ultrafiltration [48] and ion exchange chromatography [49]. Ultrafiltration has been successfully effective in removing endotoxins from water. Nevertheless, universal adoption of this technology is limited by the presence of proteins, which can be damaged by physical forces [50]. Anion exchangers, which take advantage of the negative net charge of endotoxins, have been used for endotoxin adsorption. However, when negatively charged proteins need to be decontaminated, they may co-adsorb onto the matrix and cause a significant loss of biological material. Also, net-positively charged proteins form complexes with endotoxins, causing the proteins to drag endotoxin along the column and consequently minimizing the endotoxin removal efficiency [51]. LPS removal is more efficient on cationic exchangers than on anionic exchangers. In recent years, alkanediols were shown to be effective agents for the separation of LPS from LPS-protein complexes during chromatography with ionic supports. Their effectiveness in reducing the protein complexation with LPS is dependent on (I) the size of the alkanediol, (II) the isomeric form of the alkanediol, (III) the length of the alkanediol wash, (IV) the concentration of alkanediol and (V) the type of ionic support used, cationic or anionic. Membrane-based chromatography has been successfully used for PLS separations form protein, universal adoption of this technology has not taken place because membrane chromatography is limited by the binding capacity, which is small when compared to that of bead-based columns, even though the high flux advantages provided by membrane adsorbers would lead to higher productivity [52-63].
Jann et al. [64] tested that slab-polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulfate (SDS-PAGE) can be used for the separation of LPS. Several methods have been used to separate the different subclasses of LPS from individual strains, with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and gel filtration being perhaps the most successful [65-75]. These methods are hampered by the tendency of LPS to aggregate and by the difficulty in detecting and identifying each distinct subclass [76]. Reichelt et al. [77] reported that the removal of endotoxin could be achieved during chromatography purification with the use of Triton X-114 in the washing steps. The application of 0.1% Triton X-114 in the washing steps was successful at reducing endotoxins during histidine and GST (resin GST sepharose) fusion protein purification, whereas washing steps lacking surfactant were ineffective in eliminating endotoxins. In contrast to purified materials employing the standard protocol which contained from 2500 to 34000 EU mg-1, purified recombinant proteins treated with Triton X-114 contained concentrations as low as 0.2 to 4 EU mg-1 (less than 1% of initial endotoxin content). Reichelt et al. [77] studied whether the use of Triton X-114 in washing steps could eliminate endotoxins from proteins with a pI above 8.5. They found that washing with Triton X-114 coupled with affinity chromatography effectively removed endotoxins from negatively-charged proteins (SyCRP and NdhR). The minimal endotoxin concentration achieved was lower than 0.2 EU mg-1; protein recovery and yield were close to 100% [77]. The use of two-phase aqueous micellar systems for the purification or concentration of biological molecules, such as proteins and viruses has been growing [78-80]. In these systems an aqueous surfactant solution, under the appropriate solution conditions, spontaneously separates into two predominantly aqueous, yet immiscible, liquid phases, one of which has a greater concentration of micelles than the other [81]. The difference between the physicochemical environments in the micelle-rich phase and in the micelle-poor phase forms the basis of an effective separation and makes two-phase aqueous micellar systems a convenient and potentially useful method for the separation, purification and concentration of biomaterials [82]. Particularly for endotoxin removal, above the critical micelle concentration (CMC) of surfactants, endotoxins are accommodated in the micellar structure by non-polar interactions of alkyl chains of lipid A and the surfactant tail groups and are consequently separated from the water phase (micelle-poor phase). Surfactants of the Triton series show a miscibility gap in aqueous solutions. Above a critical temperature, the so-called cloud point, micelles aggregate to droplets with very low water content, by that forming a new phase. Endotoxins remain in the surfactant-rich phase. Through centrifugation or further increase in temperature the two-phases separate with the surfactant-rich phase being the bottom phase [83,84]. If necessary, this process is repeated until the remaining endotoxin concentration is below the threshold limit. The cloud point of Triton X-114 is at 22°C, which is advantageous when purifying proteins. Adam et al. [85] used Triton X-114, showed that a 100-fold endotoxin reduction in two steps with a final endotoxin content of 30 EU mg-1 and 50% loss in bioactivity of the exopolysaccharide. In addition, about 100-fold endotoxin reduction was shown by Cotten et al. [86] from plasmid DNA preparation with a final endotoxin content of 0.1 EU in 6 μg DNA. The detergents, even though they were also very effective at reducing the LPS levels, are relatively expensive, would add significant cost to a manufacturing process and may affect the bioactivity of the protein of interest. Alternative chemicals are desired that could safely and cost effectively be used in place of the alcohols or detergents as washing agents for the separation of LPS from proteins during chromatographic unit operations [87]. Indeed, these chemicals would be relatively inexpensive, chemically well-defined, present minimal safety issues, and ideally have minimal impact on the bioactivity of the protein in question when implemented into a process.
The objective of this research is to test the synthesis and applications of a new nanocomposite polymer formed by mixing and cross-linking networks graphite oxide (GO) with with epichlorohydrin (ECH) as a coupling agent and diethylenetriamineas (DETA) a ligand. Then, we will examine the mechanical properties of the nanocomposite polymer as well as its various applications in the accumulation of PLS. The fabricated GO nanocomposites polymers have better mechanical and thermal properties and stabilities than GO nanocomposites alone. The results has shown strong interactions between the functional groups of the three components, confirmed by FTIR spectra, led to a series of improved properties, including mechanical strength in both wet and dry conditions. The results of this research has shown that the novel graphene oxide (GO)-based adsorbent embedded with epichlorohydrin (ECH) as a coupling agent and diethylenetriamineas (DETA) as a ligand (GO-ECH-DETA), has excellent endotoxin removal from aqueous solutions and biotechnological preparations. The results of our studies showed that endotoxin strongly loads on the GO-ECH- DETA nanocomposite derivative via σ–σ, π–π and n–π interaction and bonding between the nanocmposite and the LPS. The interaction and adsorption behavior of the prepared composite was elucidated with a series of experiments. The results revealed that the adsorption mechanism dominated between endotoxin molecules and the GO-ECH-DETA matrix favor acidic conditions were the optimum for the adsorption process at pH 5.5. The Langmuir–Hinshelwood kinetic model adequately describes the experimental results; both the pseudo-first order kinetic constants of the reactions and the adsorption constants were calculated. GO-ECH-DETA was more active than GO alone for the endotoxin accumulation. The reduction of 90% of the endotoxin was observed after 1hr. Thus, graphene polymer nanocomposites (GO-ECH-DETA) offer a green alternative to synthetic polymers in the preparation of soft nanomaterials, results indicated that a significant interaction of the amines of the DETA with both GO and ECH and ECH-DETA polymer were inserted between the GO layers and (ii) ECH reacted with carboxyl and epoxy groups of GO, leading to its reduction and hence the destruction of the layered structure.
MATERIALS AND METHOD
GO synthesis
GO was synthesized through a modified Hummer's method (88): 6 g of natural graphite powder (Graphene Laboratories Inc.), 4.5 g sodium nitrate and 207 mL sulphuric acid were added in a reaction flask, kept at 10°C and stirred for 30 min, followed by the addition of 27 g potassium permanganate. The solution was stirred for 45 min and then 414 mL of water was added. After 12 h, 1260 mL of warm water and 45 mL oxygen peroxide (30%) were added. The suspension was filtered, washed several times and finally dried at 60°C in a vacuum oven.
Nanocomposite fabrication
The epoxy resin used in this study was epichlorohydrin (ECH) as a coupling agent and diethylenetriamineas (DETA) a ligand and the hardener, both supplied by Huntsman. Nanocomposite samples were prepared without nanomaterials (i.e., neat epoxy resin), with GO and with DETA. They all received the same amount of filling material, 0.25% wt. and the same amount of hardener, 27% wt. To improve GO dispersion, these fillers were each mixed with acetone through bath sonication (25 kHz) for 30 min. An aqueous solution of epichlorohydrin (ECH) (5 wt%) was prepared at 100°C upon stirring for 1 h and subsequently cooled to room temperature. An aqueous solution of diethylenetriamineas (DETA) (5 wt%) was prepared at 90°C upon stirring for 2 h and subsequently cooled to room temperature. The diethylenetriamineas (DETA) a ligand hardener was added to each mixture and heated to 90°C to melt down. The chemically reduced GO dispersion was then mixed with epichlorohydrin (ECH) epoxy resin solution at 65°C and ultrasonicated (42 kHz) for another additional 1 h. The resulting mixtures were degasified at 80°C for 24 h to eliminate volatiles such as acetone and avoid bubbles in the final nanocomposites. Curing process was held in two stages: at 80°C for 1 h and at 120°C for 2 h, according to the manufacturer's instructions [88].
Endotoxin detection methods
We used Sun et al. [89] applied cysteamine-modified gold nanoparticles to detect LPS by UV-Vis spectrum and the detection limit was decreased to 3.3 × 10-10 mol/L. More facilely, a colorimetric biosensor fabricated with gold nanorods was developed and can detect LPS in the concentration range of 0.01 to 0.6 µM. Nanorods of a high aspect ratio were also demonstrated to show superiority in sensing [89]. The facile assay for the rapid visual detection of lipopolysaccharide (LPS) molecules down to the low nanomolar level by taking advantage of the electrostatic interaction between LPS molecules and cysteamine-modified gold nanoparticles (CSH-Au NPs). The large amount of negatively charged groups on the LPS molecules make LPS highly negatively charged. Thus, when modified with cysteamine, the positively charged gold nanoparticles can aggregate in the presence of trace amounts of LPS. The probe is simple, does not require any advanced instrumentation, and the limit of detection (LOD) was determined to be as low as 3.3 × 10-10 mol/L. To the best of our knowledge, it is the most sensitive synthetic LPS sensor reported so far.
Characterization techniques
Raman spectrum was used to show the graphitic ordering before and after functionalization treatments on GO and GO-ECH-DETA samples. It was acquired on a Renishaw 2000 Micro-Raman, with Ar laser (λ=514.5 nm) and range of 500-3500 cm-1 (only first order spectrum is shown in results). XPS high-resolution spectra were obtained to determine atomic composition of GO in a UNI-SPECS UHV spectrometer (5 × 10-7 Pa, hν=1253.6 eV). FT-IR was used to characterize the presence of chemical groups on GO surfaces. Infrared spectra were recorded on a Perkin-Elmer Spectrum GX, in the range of 4000-400 cm-1 with 4 cm-1 resolution, 12 scans and KBr pellet method. X-ray photoelectron spectroscopy (XPS) was employed for the analysis of the surface chemistry of GO and GO-ECH- DETA, using a SPECS system equipped with a Phoibos 150 1D-DLD analyser (Berlin, Germany) and monochromatic Al Kα X-ray source (1486.6 eV). The XPS survey-scan spectra were recorded with pass energy of 80 eV, step energy 1 eV, and dwell time 0.1 s, whereas the individual high-resolution spectra were collected with pass energy of 30 eV, step energy 0.1 eV and dwell time 0.1 s, at an electron take-off angle of 90°. A Renishaw Invia microscope (Gloucestershire, UK) with laser frequency of 514 nm was used to obtain the Raman spectra of the graphenic materials from 500 to 3500 cm-1. The information about the methods for the structural, morphological, microstructural and thermal characterization of GO and GS is displayed in the Supplementary Material. The XRD patterns of graphenic materials, GO and GO-ECH-DETA nanocomposites were performed on a Malvern Panalytical (Almelo, Netherlands) X’PERT PRO automatic diffractometer operating at 40 kV and 40 mA, in theta-theta configuration, secondary monochromator with Cu-Kα radiation (λ=0.154 nm) and a PIXcel solid state detector (active length in 2θ 3.347°). Data were collected in the range of 2θ=1-50° (step size of 0.026° and time per step of 80 s, total time 20 min) at room temperature. A variable divergence slit giving a constant 5 mm area of sample illumination was used. The Bragg equation (λ=2d sinθ) was used to determine the interlayer distance in the graphenic materials. A Hitachi S-4800 scanning electron microscope (Tokyo, Japan) operating at an accelerating voltage of 15 kV was used to obtain SEM images of the neat GO-ECH-DETA nanocomposite films, after being freeze fractured by liquid nitrogen and sputtered with gold. TEM micrographs of nanocomposites were obtained with a Philips Tecnai G2 20 TWIN TEM (Eindhoven, Netherlands) at 200 kV accelerated voltage after cutting the GO-ECH-DETA films into thin sections with a Leica EM UC6 ultramicrotome apparatus, at room temperature and placing the sliced specimens in copper grids. Differential scanning calorimetry analyses were performed by a Mettler Toledo DSC 3+ unit (Greifensee, Switzerland). The samples were heated from −30°C to 250°C at a heating rate of 10°C/min under a nitrogen gas flow of 20 mL/min. Values were obtained from the first cooling and second heating scans. Thermogravimetric analysis was performed on a TA instruments TG-Q-500 (New Castle, DE, USA) at a heating rate of 10°C/min from 40°C to 800°C in nitrogen or air-flow. An electromechanical testing machine (Instron 5967, Norwood, MA, USA) operating at room temperature with a load cell of 500 N, a gauge length of 10 mm and a cross head speed of 5 mm/min was used to performed tensile tests. Films were were cut into a dog-bone shape before testing and kept at a relative humidity of 58% at room temperature for more than one week to ensure equilibration of the moisture uptake in the films. Testing was carried out on at least ten identical composite films of each composition and the average values were reported [90].
RESULTS
X-ray diffraction (XRD) analysis
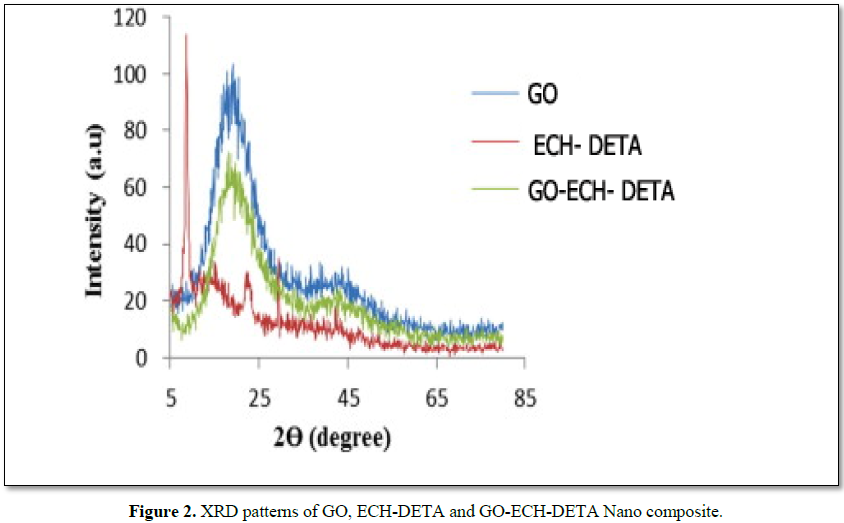
Figure 2 shows the XRD patterns of GO, ECH-DETA and GO-ECH-DETA nanocomposite The basal reflection peak (0 0 1) of pure GO at 2θ ≈ 8.00 is shifted to 2θ ≈ 20.50 in the GO-ECH-DETA spectrum because of the intercalation of ECH and DETA chains in the interlayer spacing of GO. Both pure ECH-DETA and the GO-ECH-DETA nanocomposites exhibit an intense peak at a 2θ value of 22.50, corresponding to a basal spacing of 3.95 A°. This indicates the formation of several layers of GO. The peak at 2θ ≈ 22.50 is attributable to the formation of graphene oxide structures. The broad peak centered at 2θ ≈ 22.50 is attributable to the intercalation of ECH epoxy chains between the stacked GO layers. The spectrum of GO-ECH-DETA nanocomposite shows two amorphous peaks at 2θ ≈ 20.50 and 22.50. The absence of characteristic peak of GO in the composites indicates the delamination of GO layers in the presence of ECH epoxy. The XRD data also imply the homogeneous dispersion of GO in the nanocomposites. However, XRD is not the best tool to determine crystal layer delamination or the homogeneity of dispersion. High magnification electron microscope can be used to confirm the homogeneity of the composites. The results from XRD analysis of the sample film formed has a bandwidth on the lower diffraction angle the results from XRD analysis of the sample film formed has a bandwidth on the lower diffraction angle (2θ=1.97°) corresponding to the existence of oxygen-rich groups on both sides of the leaves and the water molecule inserted between the GO sheets. The existing peaks in the diffractogram similar suggest that the ECH-DETA chains are interspersed between the GO layers. Thus, maintaining the arrangement of the graphene nanosheets, suggesting that after the functionalization of graphene oxide with the ECH and DETA the occurrence of flare spacing d is attributed to covalently bonded interconnection with the GO. In fact, the ECH-DETA is bonded on the surface of GO; can be attributed to the diffraction band orientation of GO sheets with the ECH and DETA chains crosslink's to form a GO-ECH-DETA nanocomposite lattice.SEM images
Figure 3 shows SEM images of GO before and after functionalization treatments of GO with ECH-DETA to form GO-ECH-DETA nanocomposite polymer showed a slight degree of tangling, but the blocks were clearly undone and the basic structure of the flakes was preserved. SEM images of GO and GO-ECH-DETA nanocomposite evidence the good dispersion state of graphene sheets throughout ECH-DETA. Single dispersed sheets and aggregated nanosheets with thickness ~12 nm coexist. However, a better degree of dispersion is achieved in the polymerized nanocomposites. From SEM patterns it can be inferred an exfoliated morphology for the samples prepared by the in situ method. For the nanocomposite sample obtained by the ex situ method, a good dispersion and exfoliation of graphene sheets is observed.The SEM images of tensile fracture surfaces with pure GO are shown in Figure 3. A smooth fracture surface is seen in Figure 3a at 500x magnification, which indicates a brittle fracture. Figure 3b shows how with 1.5 vol. % ECH-DETA, the fracture surface becomes cloud-like and rough. Adding more GO roughens the fracture surface. The cracks become more randomly dispersed, indicating that the GO network acts as an obstacle to crack propagation. GO has a high specific surface area. In this image the SEM of Figure 3b have a display surface in some areas of irregular morphology of fibers and various forms of holes evenly along the fiber, are structures are similar to the rod. But, there are 2 wt. % graphene oxide inclusions longer the polymer, the fibers presented in the fracture morphology is connected; indicating a high physical interaction existing between ECH-DETA and GO nanocomposite. Overall, graphene has carboxylic acid functional group provides an intermolecular force calling itself bridge effect. Providing a cohesive bond on the GO with ECH-DETA system provides a more efficient load transfer to the polymer matrix. Thus, ECH- DETA addition enhances the strength of the composite. However, further addition of ECH-DETA (beyond 1.5 vol.%) leads to the formation of clusters within the GO network that are in the scale of microns. This inhibits the stress transfer from the ECH-DETA matrix to the GO network, thus deteriorating the strength of the composite. It is clear from the SEM image that GO is generally dispersed properly in the matrix. Upon impact, the crack propagates in the direction of the tension, and then proceeds to the weak interfaces, finally damaging the material. At 4.5 vol.% ECH- DETA, the fracture surface is non-uniform. The ECH- DETA flow is hindered by the GO agglomerates. The composite surface shows signs of brittleness, with a rougher surface than neat ECH-DETA. The pull-out of the GO in the ECH-DETA matrix is also seen to decrease.The presence of functional groups GO surface was confirmed by XPS analysis. As expected, oxygen and nitrogen were found in GO exhibits high oxygen content from its oxidation. Also, exploratory scans have indicated the residual presence of sulfur from growth and functionalization processes in. In our studies, ECH-DETA was grafted onto the GO sheets by in situ ring opening polymerization of GO. The grafted GO-ECH-DETA dissolved well in dichloromethane, chloroform, DMF, THF, toluene and ethylene acetate. The homogeneous dispersion of GO in the polymer matrix improved the mechanical properties of ECH-DETA. The aggregation and stacking of GO nanosheets were also supported by tethering GO sheets on the ECH-DETA chains. We verified this morphology the presence of well-dispersed layers, indicating that after chemical reduction the material has not organized its crystal structure. Consequently, an increase in surface area of these nanoparticles in the Nanocomposite, allowing the modification of the polymeric matrix structure, and this may result in increased elastic modulus and hardness of the sample; possibly change in the degree of crystalline because smaller nanoparticles can act as nucleation sites.
XPS scanning spectrum
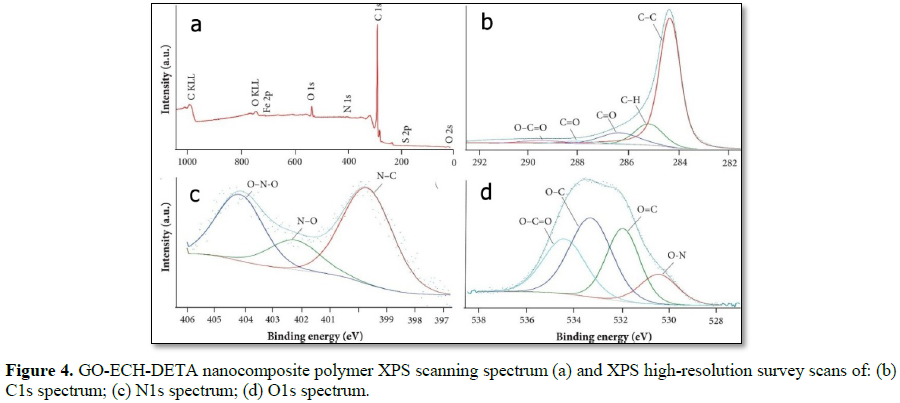
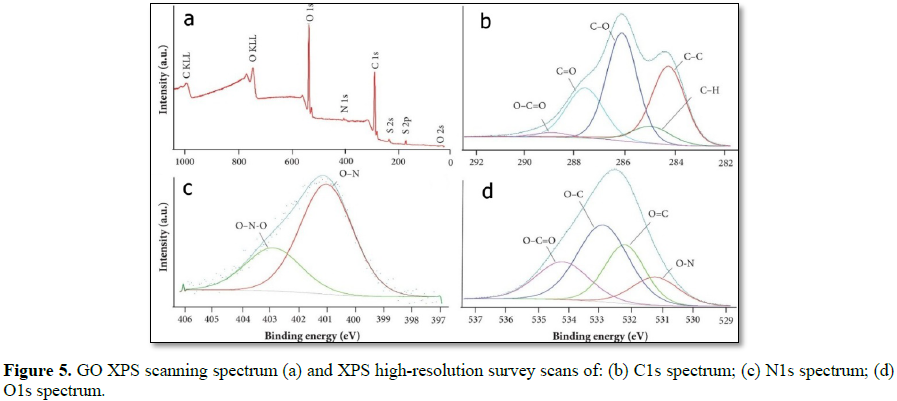
The presence of functional groups on GO surface before and after functionalization treatments of GO with ECH-DETA to form GO-ECH-DETA nanocomposite polymer surface was confirmed by XPS analysis (Table 1). As expected, oxygen and nitrogen were found in GO and GO-ECH-DETA exhibits high oxygen content from its oxidation. Also, exploratory scans have indicated the residual presence of sulphur and from growth and functionalization processes in GO-ECH- DETA, as shown in Figure 4, as well as residual presence of sulphur in GO from its synthesis process as shown in Figure 5.Raman spectrum of GO and GO-ECH-DETA nano composite
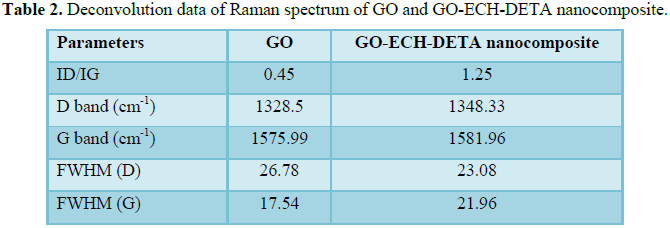
Figure 6 shows the Raman spectrum of GO and GO-ECH-DETA nanocomposite at different temperature as grown have clear bands at 1348.91 cm-1 (D band) and 1575.99 cm-1 (G band) and a shoulder near 1591.21 cm-1 (D' band). After functionalization, GO-ECH-DETA nanocomposite presents two bands at 1350.33 cm-1 (D band) and 1581.96 cm-1 (G band) and a shoulder near 1614.64 cm-1 (D' band). These bands are characteristic of multi-walled GO. The higher intensity of the G band for CNTs as grown indicates a higher degree of graphitisation/crystallinity, while D band is typically attributed to disordered structures (defective GO and non-crystalline carbon. The change in intensities of D and G band could be observed on the Raman spectrum of GO-ECH-DETA due to the acid and amino functionalization process. It is known that during oxidizing treatments of graphitic structures two concurring phenomena take place: the removal of amorphous carbon from the GO and the formation of oxygenated functional groups, changing the atomic structure from C-C sp2 to C-C sp3. Due to this change, a displacement in the position of G band and a higher intensity of D band can be observed.ID/IG ratio changed from 0.45 for GO as grown to 1.25 for functionalized GO-ECH-DETA nanocomposite. The increase in ID/IG ratio suggests that formation of oxygenated functional groups was more intense than removal of amorphous carbon.
FT-IR analysis
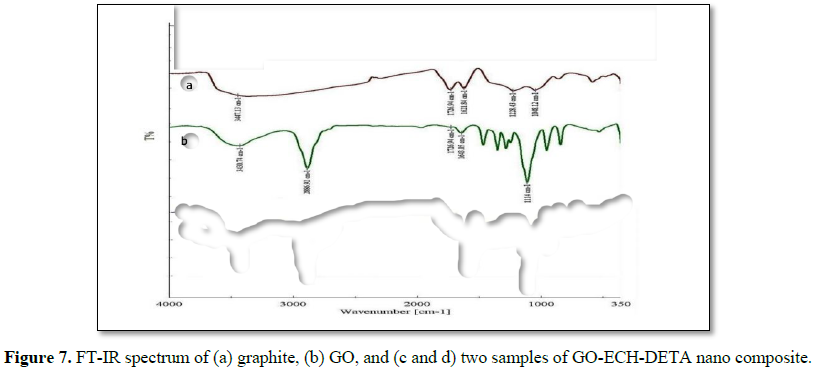
The FTIR spectra were obtained between 4000 and 350 cm-1. Figure 7 shows the FT-IR spectrum of (a) GO and (b) GO-ECH-DETA nanocomposite. The technique was used to investigate the structure and functional groups of both samples. Figure 7b shows FT-IR spectrum of GO, shows adsorption bands at 1723 cm-1, due to the C=O stretch of -COOH group; at 1621 cm-1, for stretch of C=C groups; at 1220 cm-1, for C=C skeleton vibration; and at 1043 cm-1 for alkoxy C-O groups. Although graphite had been oxidized into GO, C=C groups led to the conclusion that the main structure of graphite layer was retained. The presence of oxygen-containing functional groups confirmed that the graphite was greatly oxidized into GO and was in agreement with the literature. FT-IR spectrum for GO-ECH-DETA nanocomposite sample (Figures 7c and 7d), shows the following bands and peaks of interest: at 1670 cm-1, corresponding to the amide carbonyl (C=O) stretching; at 3728 cm-1, due to -NH stretching; at 1587 cm-1, because of N-H in-plane bending; and at 1220 and 1047 cm-1, assigned to C-N stretching. These bands prove the presence of amide groups and lead to the conclusion that carboxylic groups on GOs surface were modified by amine from DETA. The band in the range of 3445.15 cm-1 showed a relatively broad bandwidth that is probably related to the axial deformation of the O-H bond due to the reaction with ECH. The other bands in the range of 3026.35 cm-1 C-H show that the nanocomposite formation process between the ECH, DETA and GO on the surface was successful. However, the characteristics and similar bands have the predominance of GO. The oxygen atoms tend to combine with carbon atoms thereby forming an array of functionality, among which can be mentioned: ketones, esters, carboxylic acids, and others. The three faint bands were observed in the region between 1651 and 1452-1366 cm-1 due to bending vibrations and axial deformation of the C = C bonds is low because of GO with respect to the nanocomposite polymer matrix. The two most intense peaks has its stretching vibration ascribed to C = O appeared in the range 1493-1601 cm-1 are due to the formation of hydroxyl and carboxyl groups, resulting from the chemical reaction. The band located at 748 cm-1 is related to the C-axial deformation the primary alcohols and other of band and 540 cm-1 are due to the angular deflection of C-H with H out of plane. Therefore, GOs were indeed functionalized through ECH and DETA treatments.
LPS removal by GO-ECH-DETA nano composite
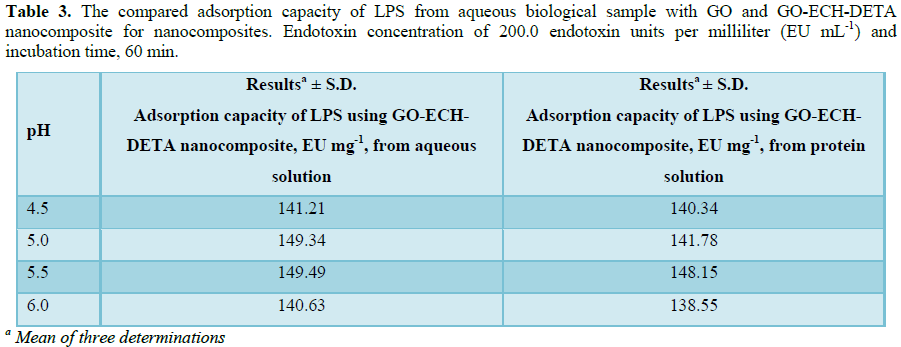
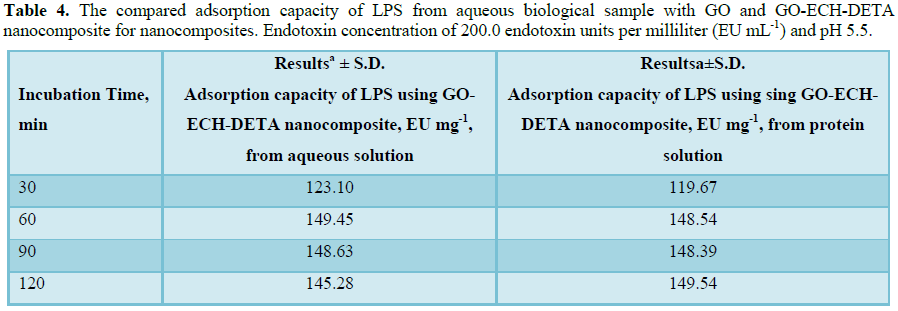
Several experiments were done to evaluate GO-ECH-DETA nanocomposite for their ability to eliminate LPS from aqueous solution and protein preparations. First, 50 ml protein solution (10 ppm) containing 200.0 endotoxin units per milliliter (EU mL-1) was subjected to dialysis for 24 h against distilled water using a Slide-A-Lyzer 10 kDa cassette (Life Technologies, cat. # 66380). Second, a volume of 1 ml of 0.1% TX-114 in buffer and 1 ml of 1.0 mg/ml of BLG were applied to a HiTrap Desalting column (GE Healthcare Life Sciences, cat. # 29-0486-84). Third, samples were centrifuged at 21,000 g and 25°C for 10 and 20 min and at 37°C for 10 and 20 min. Fourth, samples were centrifuged using 0.22 μm (cat. # 8160) and 0.45 μm (cat. # 8162) spin X filter columns (Costar) at 10,000 g at 37°C for 4 min. Last, 50 mg of GO-ECH-DETA nanocomposite were tested for their efficacy to remove LPS from both pure aqueous solution and the protein solution, at 25°C. The LPS concentration was measured using the commercially available Endozyme Recombinant Factor C assay (Hyglos, cat. # 609050) according to the protocol of the manufacturer. The measurements were conducted in triplicate using a Tecan Infinite 200Pro plate reader. The positive control (MQ) was spiked with a concentration of 0.45 EU/l of LPS, and results for all tests were considered valid when the value of recovered LPS concentration was between 50 and 200% of this value. LPS units were converted from EU (Endotoxin Unit) to concentrations in pg/ml by assuming that 1 EU corresponds to 100 pg of standard endotoxin EC-5. Thus, this is a simple and cheap procedure, has proven to remove endotoxins without affecting any significant losses in protein yields and biological activities. The initial concentration of LPS in both pure aqueous solution and the protein solution was estimated to be 150 EU/ml. GO-ECH-DETA nanocomposite LPS extraction reduced the LPS concentration in biological protein sample to 1.85 EU/ml and aqueous solution to 0.51, demonstrating in purification efficiencies of 98.58% and 99.7%, respectively (Tables 3 and 4). Repeated (one, two or three times) LPS-extraction did not lower LPS levels further.CONCLUSION
In summary, we developed multi-functional GO-based nanocomposites, GO-ECH-DETA, by the reaction of graphite oxide (GO) with epichlorohydrin (ECH) as a coupling agent and diethylenetriamineas (DETA) a ligand. The instrumental analysis of the nanocomposite prove that there is chemical interaction between ECH, DETA and GO on the surface of GO. For example, the presence of amide groups and lead to the conclusion that carboxylic groups on GOs surface were modified by amine from DETA. The band in the range of 3445.15 cm-1 showed a relatively broad bandwidth that is probably related to the axial deformation of the O-H bond due to the reaction with ECH. The other bands in the range of 3026.35 cm-1 C-H show that the nanocomposite formation process between the ECH, DETA and GO on the surface was successful. The polymeric chains on the surface of GO endowed GO-ECH-DETA nanocomposite with excellent chain stability. Additionally, from the SEM images we observed the interface between the GO and the epoxy composite ECH-DETA suggests chemical interaction. As can be seen from this research, the GO-ECH-DETA nanocomposite can be used as a potential candidate for removal of PLS from aqueous solution.
CONFLICTS OF INTEREST
The authors declare no conflict of interest regarding the publication of this paper.1. Gorbet MB, Sefton MV (2014) Endotoxin: The uninvited guest. Biomaterials 26: 6811-6817.
2. Ryan J (2014) Endotoxins and cell culture. Corning Life Sciences Technical Bulletin 1: 1-8.
3. Koyuncu I, Arikan OA, Wiesner MR, Rice C (2008) Removal of hormones and antibiotics by nano filtration membranes. J Membrane Sci 309: 94-101.
4. Anspach FB (2001) Endotoxin removal by affinity sorbents. J Biochem Biophys Methods 49: 665-681.
5. Erridge C, Bennett-Guerrero E, Poxton IR (2002) Structure and function of lipopolysaccharides. Microbes Infect 4: 837-851.
6. Ogikubo Y, Ogikubo Y, Norimatsu M, Noda K, Takahashi J, et al. (2004) Evaluation of the bacterial endotoxin test for quantification of endotoxin contamination of porcine vaccines. Biologicals 32: 88-93.
7. Fiske JM, Ross A, VanDerMeid RK, McMichael JC, Arumugham (2001) Method for reducing endotoxin in Moraxella catarrhalis UspA2 protein preparations. J Chromatogr B Biomed Sci Appl 753: 269-278.
8. Daneshiam M, Guenther A, Wendel A, Hartung T, Von Aulock S (2006) In vitro pyrogen test for toxic or immunomodulatory drugs. J Immunol Methods 313: 169-175.
9. Ohno N, Morrison DC (1989) Lipopolysaccharide interaction with lysozyme: Binding of lipopolysaccharide to lysozyme and inhibition of lysozyme enzymatic activity. J Biol Chem 264: 4434-4441.
10. Bennett IL Jr, Beeson PB, Roberts E (1953) Studies on the pathogenensis of fever: The effect of injection of extracts and suspensions of unifected rabbit tissues upon the body temperature of normal rabbits. J Exp Med 98: 477-492.
11. Hirayama C, Sakata M (2002) Chromatographic removal of endotoxin from protein solutions by polymer particles. J Chromatogr B Analyt Technol Biomed life Sci 781: 419-432.
12. Berthold W, Walter J (1994) Protein purification: Aspects of processes for pharmaceutical products. Biologicals 22: 135-150.
13. Petsch D, Anspach FB (2000) Endotoxin removal from protein solutions. J Biotechnol 76: 97-119.
14. Lin MF, Williams C, Murray MV, Ropp PA (2005) Removal of lipopolysaccharides from protein-lipopolysaccharide complex by nonflammable solvents. J Chromatogr B Analyt Technol Biomed Life Sci 816: 167-174.
15. Hoffmann S, Peterbauer A, Schindler S, Fennrich S, Poole S, et al. (2005) International validation of novel pyrogen testes based on human monocytoid cells. J Immunol Methods 298: 161-173.
16. Ding JL, Ho BA (2001) New era in pyrogen testing. Trends Biotechnol 19: 277-281.
17. Ong KG, Leland JM, Zeng KF, Barrett G, Zourob M, et al. (2006) A rapid highly-sensitive endotoxin detection system. Biosens Bioelectron 21: 2270-2274.
18. Sullivan JD, Watson SW (1974) Factors affecting the sensitivity of limulus lysate. Appl Microbiol 28: 1023-1028.
19. Haishima Y, Hasegawa C, Yagami T, Tsuchiya T, Matsuda R, et al. (2003) Estimation of uncertainty in kinetic-colorimetric assay of bacterial endotoxins. J Pharm Biomed Anal 32: 495-503.
20. Webster CJ (1980) Principles of a quantitative assay for bacterial endotoxins in blood that uses limulus lysate and a chromogenic substrate. J Clin Microbiol 12: 644-650.
21. Poole S, Mistry Y, Ball C, Das REG, Opie LP, et al. (2003) A rapid ‘on-plate’ in vitro test for pyrogens. J Immunol Methods 274: 209-220.
22. Han D, Yan L, ChenW, Li W (2011) Reparation of chitosan/graphene oxide composite film with enhanced mechanical strength in the wet state. Carbohydrate Polymers 83: 653-658.
23. Pati MK, Pattojoshi P, Roy GS (2015) Fabrication and characterization of graphene based nanocomposite for electrical properties. Adv Mater Phys Chem 5: 22-30.
24. Devi R, Relhan S, Pundir CS (2013) Construction of a chitosan/polyaniline/graphene oxide nanoparticles/polypyrrole/Au electrode for amperometric determination of urinary/plasma oxalate. Sens Actuators B Chem 186: 17-26.
25. Li X, Zhou H, Wu WM Wei S, Xu Y, et al. (2015) Studies of heavy metal ion adsorption on chitosan/sulfydryl-functionalized graphene oxide composites. J Colloid Interface Sci 448: 389-397.
26. Layek RK, Samanta S, Nandi AK (2012) Graphene sulphonic acid/chitosan nano biocomposites with tunable mechanical and conductivity properties. Polymer 53: 2265-2273.
27. Jagiello J, Judek J, Zdrojek M, Aksienionek M, Lipinska L (2014) Production of graphene composite by direct graphite exfoliation with chitosan. Mater Chem Phys 148: 507-511.
28. Krishnan D, Kim F, Luo J, Cruz-Silva R, Cote LJ, et al. (2012) Energetic graphene oxide: Challenges and opportunities. Nano Today 7: 137-152.
29. Singh V, Joung D, Zhai L, Das S, Khondaker SI, et al. (2011) Graphene based materials: Past, present and future. Prog Mater Sci 56: 1178-1271.
30. Malafaya PB, Silva GA, Reis RL (2007) Natural-origin polymers as carriers and scaffolds for biomolecules and cell delivery in tissue engineering applications. Adv Drug Deliv Rev 59: 207-233.
31. Shelke NB, James R, Laurencin CT, Kumbar SG (2014) Polysaccharide biomaterials for drug delivery and regenerative engineering. Polym Adv Technol 25: 448-460.
32. Kyzas GZ, Deliyanni EA, Matis KA (2014) Graphene oxide and its application as an adsorbent for wastewater treatment. J Chem Technol Biotechnol 89: 196-205.
33. Travlou NA, Kyzas GZ, Lazaridis NK, Deliyanni EA (2013) Graphite oxide/chitosan composite for reactive dye removal. Chem Eng J 217: 256-265.
34. Travlou NA, Kyzas GZ, Lazaridis NK, Deliyanni EA (2013) Functionalization of graphite oxide with magnetic chitosan for the preparation of a nanocomposite dye adsorbent. Langmuir 29: 1657-1668.
35. Kyzas GZ, Travlou NA, Deliyanni EA (2014) The role of chitosan as nanofiller of graphite oxide for the removal of toxic mercury ions. Colloids Surf B Biointerfaces 113: 467-476.
36. Kyzas GZ, Bikiaris DN, Seredych M, Bandosz TJ, Deliyanni EA (2014) Removal of dorzolamide from biomedical wastewaters with adsorption onto graphite oxide/poly(acrylic acid) grafted chitosan nanocomposite. Bioresour Technol 152: 399-406.
37. Du J, Cheng HM (2012) The fabrication, properties and uses of graphene/polymer composites. Macromol Chem Phys 213: 1060-1077.
38. Das TK, Prusty S (2013) Graphene-based polymer composites and their applications. Polym Plast Technol Eng 52: 319-331.
39. Sun X, Sun H, Li H, Peng H (2013) Developing polymer composite materials: Carbon nanotubes or grapheme. Adv Mater 25: 5153-5176.
40. Kim H, Abdala AA, Mac Osko CW (2010) Graphene/polymer nanocomposites. Macromolecules 43: 6515-6530.
41. Huang X, Qi X, Boey F, Zhang H (2012) Graphene-based composites. Chem Soc Rev 41: 666-686.
42. Zhang X, Rajaraman BRS, Liu H, Ramakrishna S (2014) Graphene’s potential in materials science and engineering. RSC Adv 4: 28987-29011.
43. Tjong SC (2014) Polymer composites with graphene nanofillers: Electrical properties and applications. J Nanosci Nanotechnol 14: 1154-1168.
44. Kuang D, Hu WB (2013) Research progress of graphene composites. Wuji Cailiao Xuebao. J Inorg Mater 28: 235-246.
45. Rinaudo M (2008) Main properties and current applications of some polysaccharides as biomaterials. Polym Int 57: 397-430.
46. Draget KI, Skjåk-Bræk G, Smidsrød O (1997) Alginate based new materials. Int J Biol Macromol 21: 47-55.
47. Hote P, Miah M, Gupta A, Chkaravorty D (2019) Epichlorohydrin functionalized graphene oxide for superior Li+ ion conduction and supercapacitor application. Materials Chemistry and physics 223: 447-455.
48. Sweadner KJ, Forte M, Nelsen LL (1977) Filtration removal of endotoxin (pyrogens) in solution in different states of aggregation. Appl Environ Microbiol 34: 382-385.
49. Shibatani T, Kakimoto T, Chibata I (1983) Purification of high molecular weight urokinase from human urine and comparative study of two active forms of urokinase. Thromb Haemost 28: 91-95.
50. Pyo SH, Lee JH, Park HB, Hong SS, Kim JH (2001) A large-scale purification of recombinant histone H1.5 from Escherichia coli. Protein Expr Purif 23: 38-44.
51. Issekutz AC (1983) Removal of gram-negative endotoxin from solutions by affinity chromatography. J Immunol Methods 61: 275-281.
52. Morrison DC, Jacobs DM (1976) Binding of polymyxin B to the lipid A portion of bacterial lipopolysaccharides. Immunochemistry 13: 813-818.
53. McNeff C, Zhao Q, Almlof E, Flickinger M, Peter WC (1999) The efficient removal of endotoxins from insulin using quaternized polyethyleneimine-coated porous zirconia. Anal Biochem 274: 181-187.
54. Rao CS (2001) Purification of large proteins using ion-exchange membranes. Process Biochem 37: 247-256.
55. Gerstner JA, Hamilton SM (1992) Cramer. Membrane chromatographic systems for high-throughput protein separations. J. Chromatogr 596: 173-180.
56. Tennikova TB, Belenkii BG, Svec F (1990) High-performance membrane chromatography: A novel method of protein separation. J Liq Chromatogr 13: 63-70.
57. Tennikov MB, Gazdina NV, Tennikova TB, Svec F (1998) Effect of porous structure of macroporous polymer supports on resolution in high-performance membrane chromatography of proteins. J. Chromatogr A 6: 55-64.
58. Tennikova TB, Svec F (1993) High-performance membrane chromatography – highly efficient separation method for proteins in ion-exchange, hydrophobic interaction and reversed-phase modes. J Liq Chromatogr 646: 279-288.
59. Sarfert FT, Etzel MR (1997) Mass transfer limitations in protein separations using ion-exchange membranes. J Chromatogr A 7: 3-20.
60. Suen SY, Etzel MR (2002) Sorption kinetics and breakthrough curves for pepsin and chymosin using pepstatin A affinity membranes. J Chromatogr A 2: 179-192.
61. Teeters MA, Root TW, Lightfoot EN. Performance and scale-up of adsorptive membrane chromatography. J Chromatogr A 25: 129-139.
62. Knudsen HL, Fahrner RL, Xu Y, Norling LA, Blank GS (2001) Membrane ion-exchange chromatography for process-scale antibody purification. J Chromatogr A 12: 145-154.
63. Van Reis R, Zydney A (2001) Membrane separations in biotechnology. Curr Opin Biotechnol 12: 208-211.
64. Charlton HR, Relton JM, Slater KHN (1999) Characterisation of a generic monoclonal antibody harvesting system for adsorption of DNA by depth filters and various membranes. Bioseparation 8: 281-291.
65. Jann B, Reske K, Jann K (1975) Heterogeneity of lipopolysaccharides. Analysis of polysaccharide chain lengths by sodium dodecylsulfate-polyacrylamide gel electrophore. Eur J Biochem 60: 239-246.
66. Tsai CM, Frasch CE (1982) A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal Biochem 119: 115-119.
67. McIntire FC, Sievert HW, Barlow GH, Finley RA, Lee AY (1967) Chemical, physical, biological properties of a lipopolysaccharide from Escherichia coli K-235. Biochemistry 6: 2363-2372.
68. Oroszlan SI, Mora PT (1963) Dissociation and reconstitution of an endotoxin. Biochem Biophys Res Commun 12: 345-349.
69. Weiser MM, Rothfield L (1968) The reassociation of lipopolysaccharide, phospholipid and transferase enzymes of the bacterial cell envelope. Isolation of binary and ternary complexes. J Biol Chem 243: 1320-1328.
70. Ribi E, Anacker RL, Brown R, Haskins WT, Malmgren B, et al. (1966) Reaction of endotoxin and surfactants I. Physical and biological properties of endotoxin treated with sodium deoxycholate. J Bacteriol 92: 1493-1509.
71. Hannecart-Pokorni E, Dekegel D, Depuydt F (1973) Macromolecular structure of lipopolysaccharides from gram-negative bacteria. Eur J Biochem 38: 6-13.
72. McIntire FC, Barlow G, Sievert H, Finley R, Yoo A (1969) Studies on a lipopolysaccharide from Escherichia coli. Heterogeneity and mechanism of reversible inactivation by sodium deoxycholate. Biochemistry 8: 4063-4067.
73. Kim JS, Reuhs BL, Rahman MM, Ridley B, Carlson RW (1996) Separation of bacterial capsular and lipopolysaccharides by preparative electrophoresis. Glycobiology 6: 433-437.
74. Morrison DC, Leive L (1975) Fractions of lipopolysaccharide from Escherichia coli O111:B4 prepared by two extraction procedures. J Biol Chem 250: 2911-2919.
75. Maccari F, Volpi N (2003) Detection of submicrogram quantities of Escherichia coli lipopolysaccharides by agarose gel electrophoresis. Anal Biochem 322: 185-189.
76. Dietrich CP, Dietrich SMC (1985) Electrophoretic behaviour of acidic mucopolysaccharides in diamine buffers. Anal Biochem 701: 645-647.
77. Bianchini P, Osima B, Parma B, Dietrich CP, Takahashi HK, et al. (1985) Structural studies and “in vivo” and “in vitro” pharmacological activities of heparin fractions and fragments prepared by chemical and enzymic depolimerization. Thromb Res 40: 49-58.
78. Reichelt P, Schwarz C, Ponzeau M (2006) Single step protocol to purify recombinant proteins with low endotoxin contents. Protein Expr Purif 46: 483-488.
79. Volpi N (1994) Fractionation of heparin, dermatan sulfate and chondroitin sulfate by sequential precipitation: A method to purify a single glycosaminoglycan species from a mixture. Anal Biochem 218: 382-391.
80. Liu CL, Kamei DT, King JA, Wang DIC, Blankschtein D (1998) Separation of proteins and viruses using two-aqueous micellar systems. J Chrom B 711: 127-138.
81. Rangel-Yagui CO, Lam H, Kamei DT, Wang DIC, Pessoa-Jr A, et al. (2003) Glucose-6-phosphate dehydrogenase partitioning in two-phase aqueous mixed (nonionic/cationic) micellar systems. Biotechnol Bioeng 82: 445-456.
82. Mazzola PG, Lam H, Kavoosi M, Haynes CA, Pessoa Jr A, et al. (2006) Affinity-tagged green fluorescent protein (GFP) extraction from a clarified E. coli cell lysate using a two-phase aqueous micellar system. Biotechnol Bioeng 93: 998-1004.
83. Nikas YJ, Liu CL, Srivastava T, Abbott NL, Blankschtein D (1992) Protein partitioning in two-phase aqueous non-ionic micellar solutions. Macromolecules 25: 4794-4806.
84. Liu CL, Nikas YJ, Blankschtein D (1996) Novel bioseparations using two-phase aqueous micellar systems. Biotechnol Bioeng 52: 185-192.
85. Bordier C (1981) Phase separation of integral membrane proteins in Triton X-114 solution. J Biol Chem 256: 1604-1607.
86. Adam O, Vercellone A, Paul F, Monsan PF, Puzo G (1995) A non-degradative route for the removal of endotoxin from exopolysaccharides. Anal Biochem 225: 321-327.
87. Cotten M, Baker A, Saltik M, Wagner E, Buschle M (1994) Lipopolysaccharide is a frequent contaminant of plasmid DNA preparations and can be toxic to primary human cells in the presence of adenovirus. Gene Ther 1: 239-246.
88. Cunha T, Aires-Barros R (2002) Large-scale extraction of proteins. Mol Biotechnol 20: 29-40.
89. Wu ZS, Ren W, Gao L, Liu B, Jiang C, et al. (2009) Synthesis of high-quality graphene with a pre-determined number of layers. Carbon 47: 493-499.
90. Sun J, Ge J, Liu W (2012) A facile assay for direct colorimetric visualization of lipopolysaccharides at low nanomolar level. Nano Res 1: 1-8.
-
 Table 1
Table 1 -
Table 2
-
Table 3
-
Table 4



QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Food and Nutrition-Current Research (ISSN:2638-1095)
- Journal of Microbiology and Microbial Infections (ISSN: 2689-7660)
- Advances in Nanomedicine and Nanotechnology Research (ISSN: 2688-5476)
- Journal of Genetics and Cell Biology (ISSN:2639-3360)
- Journal of Biochemistry and Molecular Medicine (ISSN:2641-6948)
- Journal of Astronomy and Space Research
- Journal of Agriculture and Forest Meteorology Research (ISSN:2642-0449)


