3153
Views & Citations2153
Likes & Shares
Objective: The objective of this protocol is to define the procedure for process validation and to establish documented evidence that the manufacturing process is in state of control. Review the definition and types of validation. Understand the requirements for documentation and key stages in process validation. It is essential part of GMP. Definition of desirable attributes of the drug product. Determination of the controls or testing parameters that will be measured or tested.
Method: Concurrently 3 batches were taken and all critical parameters evaluated for fixing optimum process parameters for process validation.
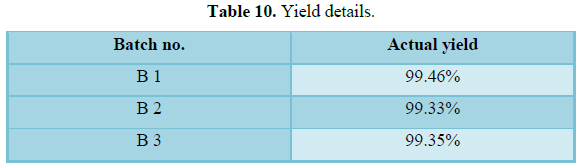
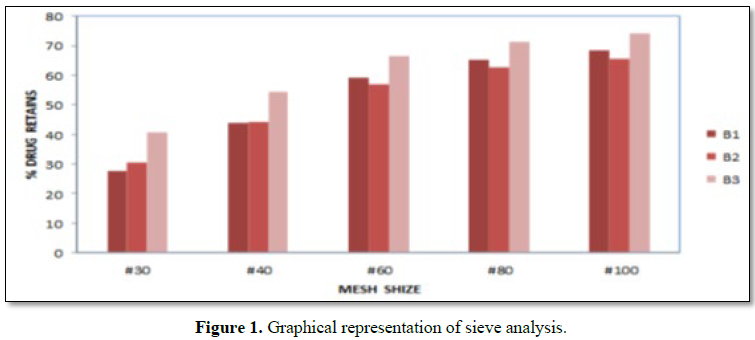
Results: The risk assessment was done for each step, and the critical parameters were validated. All the tests was found to be within the limits, and validated. Physicochemical parameter of tablets compressed with granules obtained at final impeller amperage of 11.5 to 12.5 amps, which comply with specification. The parameters in granulation stage are suggested for binder addition time, kneading time and discharge time. In the coating process all the parameters in critical steps were found within the specified limits. The sieve analysis was done for all the three batches. The sieve used and % retains are found to be within the specified limits. In the hopper study, all the parameters were found to be within the specified limits and hence the critical steps were validated. The dissolution studies for all three batches and it complies with the specification.
Conclusion: The manufacturing of three batches of common blend for Lacidipine tablets 6 mg was conducted for a batch size of 94.50 kg (210,000 tablets). The study involved validating the process variables of this transferred product to show that the process is under control. The study includes the validation of critical steps of manufacturing such as blending, drying, granulation, compression and coating. The process validation of Lacidipine tablets showed that there was no significant batch-to-batch variation. Therefore it can be concluded that the process stands validated and the data can be used in regulatory submission for obtaining marketing authorization for the Lacidipine tablets.
INTRODUCTION
The basic principle of quality assurance is that a drug should be produced that is fit for its intended use. In order to meet this principle, a good understanding of process and their performance is important. Quality should be built into the manufacturing process. These processes should be controlled in order that the finished product meets all quality specifications [30].
DEFINITION OF VALIDATION
WHO (World Health Organization)
The validation in the same way but elaborates considerably on the concept “Validation studies are essential part of good manufacturing practice and should be conducted in according with predefined protocols [30]. A Written report summarizing results and conclusions should be recorded, prepared and stored. Process and procedures should be established based upon the validation study and undergo periodic revalidation to ensure that they remain capable of achieve the intended results [31]. Particular attention should be accorded to the validation of processing, testing and cleaning procedures. Critical process should be validated, prospectively or retrospectively. When any new master formula or method of preparation is adopted, steps should be taken to demonstrate its stability for routine processing [2]. The defined process, using the materials and equipment specified should be shown to yield a product consistently of the required quality [3]. Significant amendments to the manufacturing process, including any change in equipment or materials, which may affect product quality and or reproducibility of the process, should be validated [7].
Why to validate the processes?
There are many reasons in addition to the regulatory requirements for validating processes. A manufacturer can assure through careful design of the device and packaging careful design and validation of processes and process controls that there is a high probability that all manufactured units will meet specifications and have uniform quality [11]. The dependence on intensive in-process and finished device testing can be reduced [4]. However, in-process and finished product testing still play an important role in assuring that products meet specifications. A properly validated and controlled process will yield little scrap or rework resulting in increased output. Consistent conformance to specifications is likely to result in fewer complaints and recalls. Also whenever needed the validation file will contain data to support improvements in the process or the development of the next generation of the process [5].
IMPORTANCE OF PROCESS VALIDATION
1) Reduction of Quality cost 2) Process optimization 3) Assurance of quality 4) Safety
Validation protocol
Definition: A document stating how validation will be conducted, including test parameters, product characteristics, manufacturing equipment and decision points on what constitutes acceptable test results [11].
Contents of validation protocol: 1) General information; 2) Objective, Label claim; 3) List of equipment and their qualification status; 4) Facilities qualification; 5) Process flow charts; 6) Manufacturing procedure narrative; 7) List of critical processing parameters and critical recipients; 8) Sampling, tests and specifications; 9) Acceptance criteria [17].
Process validation lifecycle
Process design: GMP, requirements for process design: 1) Design of facility; 2) Design of equipment; 3) Design of production and control procedures; 4) Design of laboratory controls; 5) Propose process steps (unit operations) and process variables (operating parameters) that need to be studied; 6) Identify sources of variability each unit operation is likely to encounter; 7) Consider possible range of variability for each input into the operation; 8) Evaluate process steps and variables for potential criticality; 9) Select process steps and variables for test in representative models; 10) Development studies to identify critical operation parameters and operating ranges; 11) Designed experiments; 12) Lab scale, pilot scale and/or full scale experimental batches to gain process understanding; 13) Establish mechanisms to limit or control variability based on experimental data; 14) Aim for a “robust process”, i.e., one that can tolerate input variability and still produce consistent acceptable output [12].
Confirmation of process: 1) Transfer developmental knowledge to Production, i.e., technology transfer; 2) Batch record and operating SOPs in place, equipment and facilities equivalency established; 3) Raw materials approved; 4) Measurement systems qualified (QC lab as well as production floor test instrumentation); 5) Personnel training completed; 6) Environment controlled as necessary; 7) Execution of confirmed batches with appropriate sampling points and sampling level; 8) First evidence that process can function at commercial scale by production personnel; 9) Demonstrates reproducibility [8].
Types of process validation
A) Prospective Validation
B) Concurrent Validation
C) Retrospective Validation
D) Revalidation
E) Periodic revalidation
Phases in process validation
A) Phase 1. (Pre-validation phase)
B) Phases 2 (Pre-validation phase)
C) Phase 3 (Process validation phase/Process qualification phase)
D) Phase 4 (Validation maintenance phase)
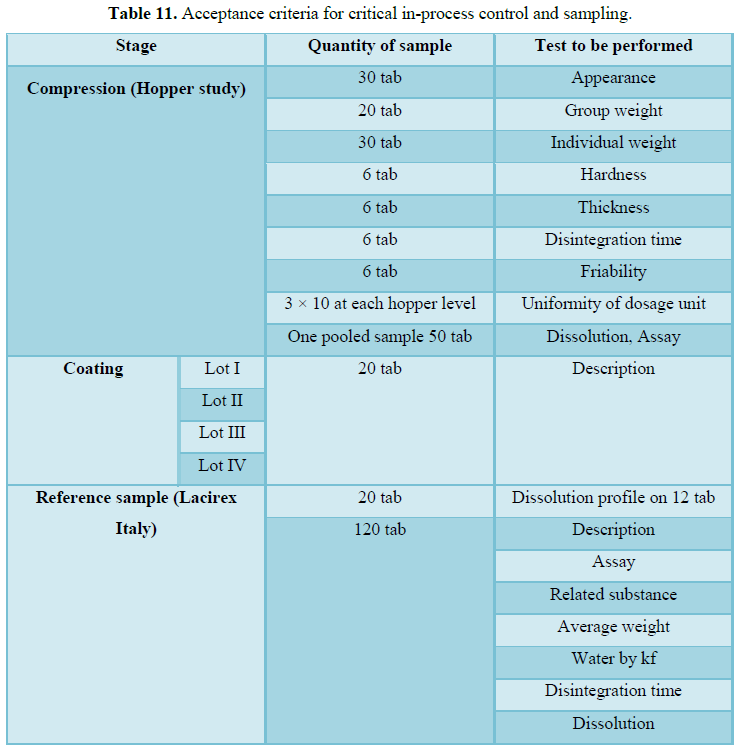
In-process quality control test includes
1) Uniformity of weight 2) Uniformity of content 3) Disintegration time 4) Friability
PROCESS PARAMETERS FOR STANADARDIZATION
Granulation
These variables affect the: 1. Granule strength; 2. Bulk density of blend; 3. Flow characteristics of granules [26].
Semi-drying and milling
1) Dust free; 2) Round, uniform shape; 3) Good flow behavior; 4) Easy to dose; 5) Good dispersibility; 6) Good solubility; 7) Compact structure; 8) Low hygroscopicity; 9) High bulk density; 10) Dense surface; 11) Narrow grain size distribution; 12) Low abrasion; 13) Visual attractiveness [9].
Drying
Moisture content in granules which determined in terms of LOD is important factor. If moisture content is more in granules it will lead to poor flow and sticking problem. If moisture is less it will lead to capping, high friability and chipping. During drying the desired LOD will be maintained in the granules which will influence the quality parameters like flow properties of granules, physical properties during compression like tablet hardness. Inlet temperature of FBD is most critical variable for the same. LOD is checked periodically to establish the same during drying [12].
Blending
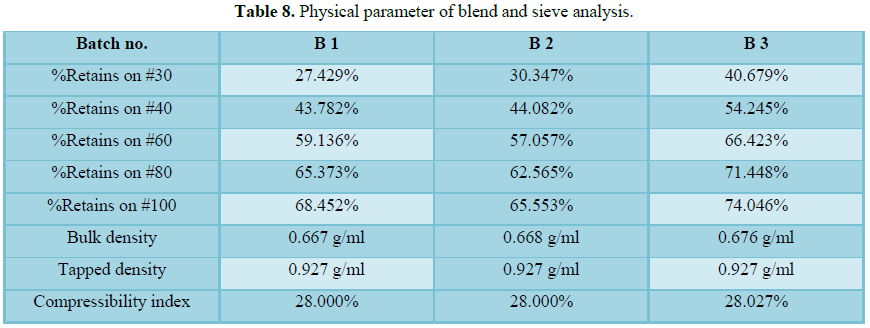
1. Bulk Density; 2. Angle of Repose; 3. Sieve analysis; 4. Compressibility Index.
Compression
Following physical parameters are to be checked to establish the above-mentioned variables at regular intervals. 1. Appearance; 2. Individual Weight variation; 3.Group Weight variation; 4. Hardness; 5. Thickness; 6. Friability [16].
Film coating
The Eight Critical Parameters for film Coating: 1. Gun geometry; 2. Automising/pattern air; 3. Pan pressure; 4. Pan speed; 5. Spray rate; 6. Inlet outlet air temperature; 7. Total air volume; 8. Adhesion of particles to the gun surface [13].
Packing
Following parameters influences speed of the machine: 1. Proper forming of blister pockets; 2. Proper sealing of blister pack; 3. Configuration of blister pack.
MATERIALS AND METHODS
Materials
Name: LACIDIPINE
Classification: Belongs to the class of dihydropyridine derivative selective calcium-channel blockers with mainly vascular effects.
Categories: Calcium antagonist.
Weight: 422.911 g/mol
Chemical formula: C26H33NO6
IUPAC name: 3,5-diethyl4-{2-[(1E)-3-(tert-butoxy)-3-oxoprop-1-en-1-yl]phenyl}-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate
Absorption: Well absorbed, the systemic bioavailability of Lacidipine is approximately 33%.
Protein binding: >95%
Metabolism: Extensive first pass metabolism.
Route of elimination: The metabolites are mainly eliminated by the biliary route and excreted via the feces.
Half-life: 13 to 19 h.
Plasma concentration: 1.6 to 5.7 μg/L
Toxicity: Hypotension and tachycardia; Bradycardia could occur from parasympathetic (vagal) stimulation, LD50=1000 mg/kg (orally in rat).
Bioavailability: 2 to 52%
Melting point: 183.5-184.5°C
State: Solid
Water solubility: 0.82 mg/L
Methods
Procurement and authentication of drug Lacidipine under study: Evaluation of three batches considering parameters listed below for I.P.Q.C tests.
1. Optimization of granulation end points
2. Evaluation of granules
3. Compression of granules into tablet
4. Evaluation of Tablet
5. Film coating of compressed tablets
6. Evaluation of coated tablets
7. Sampling of strips
8. Preparation of the validation report.
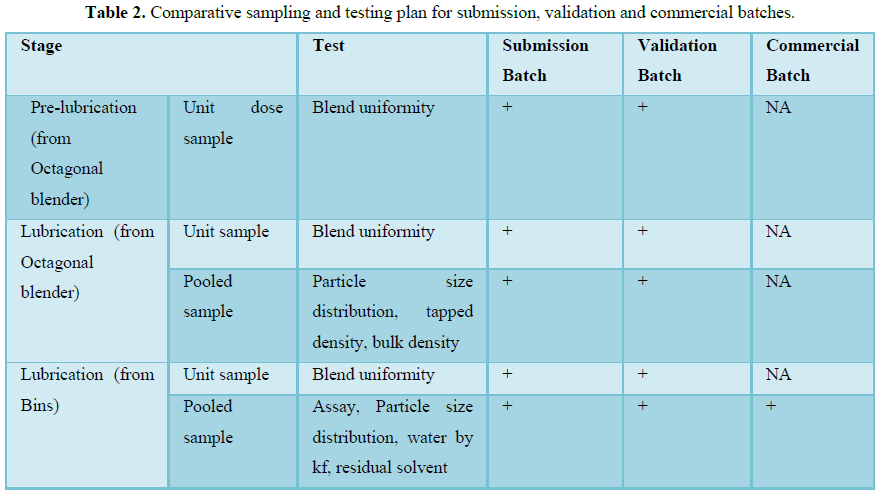
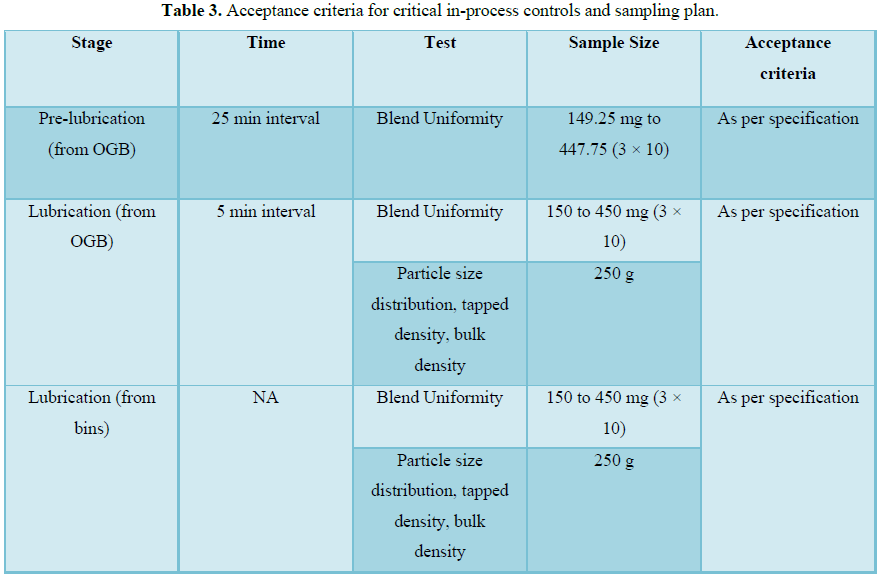
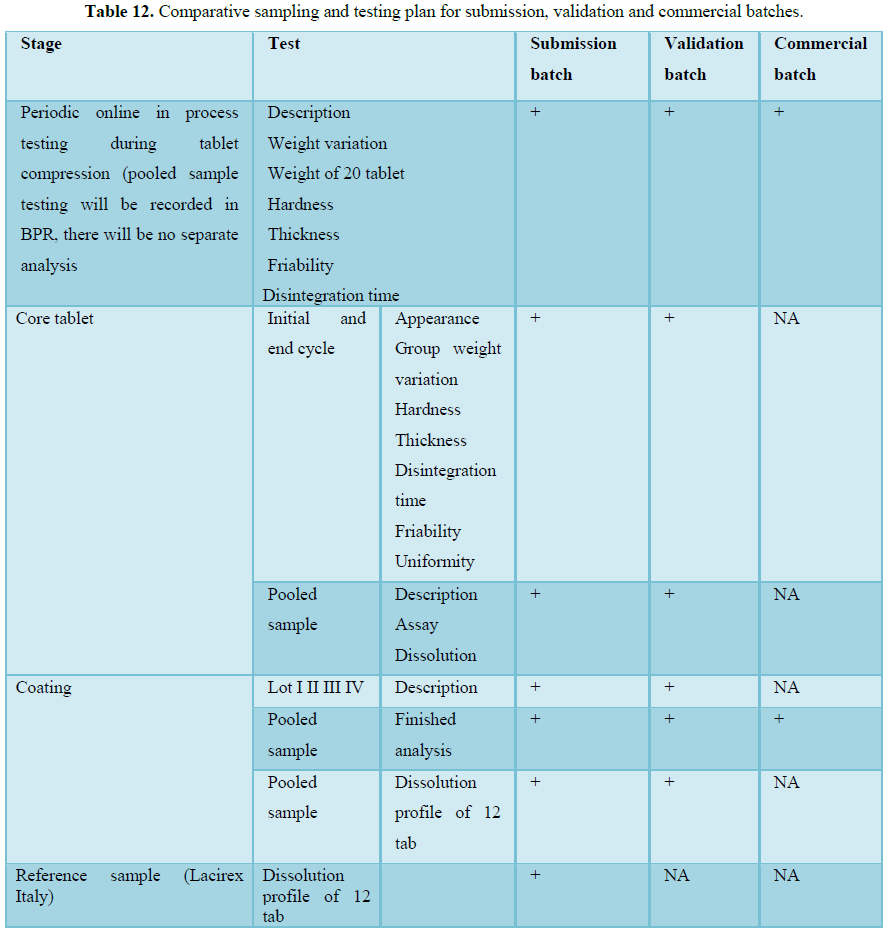
RESULTS (FIGURES 1 AND 2 AND TABLES 1-15)
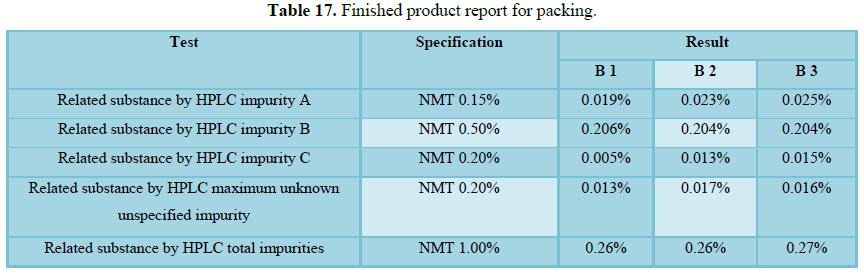
Finished product report
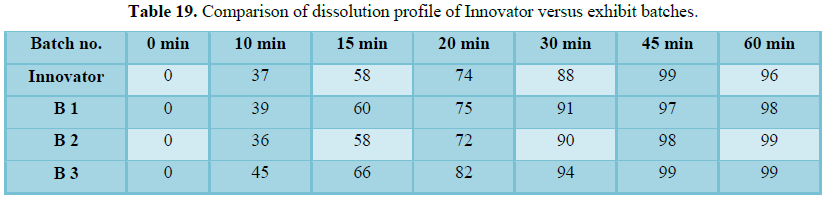
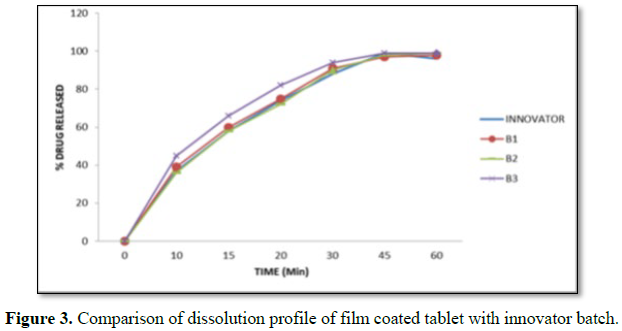
The finished product report for all the three batches was collected. All the tests for finished product were passed as per the specification (Figure 3 and Tables 16-19).
DISCUSSION
The common blend 94.50 kg was divided into three different strengths viz. 19.50/130,000 tab for 2 mg strength 30.00 kg/100,000 tab for 4 mg and 45.00 kg/100,000 tab for 6 mg tab.
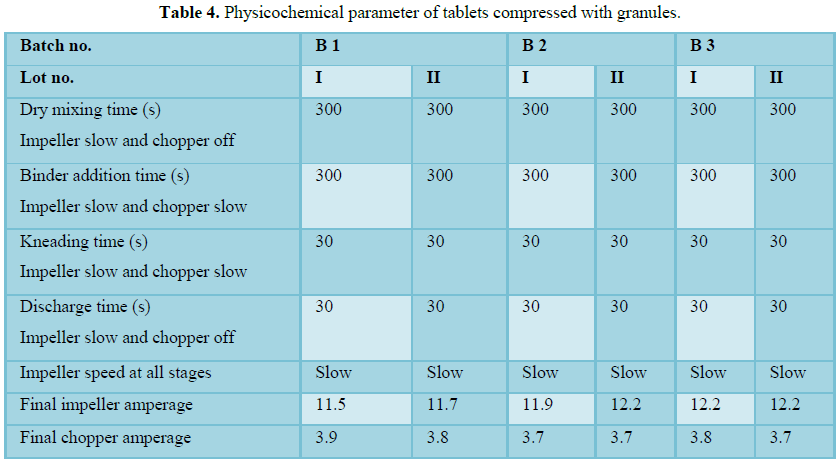
Dry mixing and granulation
Dry mix was done for 5 min at impeller slow speed (75 rpm) to match Froude number with tablet batches. Granulation was carried out at slow speed of impeller and chopper slow speed with addition of granulating solution as per manufacturing instruction which produced satisfactory granules so the binder addition time and kneading time is recommended as mentioned in manufacturing instructions.
Wet milling
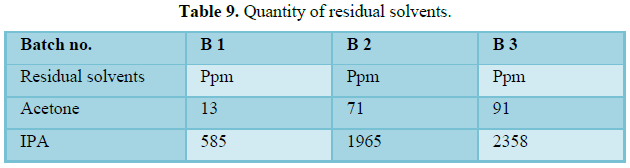
Wet milling was done in Quadro co-mill using 250Q screen to break wet mass and facilitate uniform drying to keep residual solvents within specified limits.
Drying
Drying was carried out at controlled inlet temperature of 55°C and desired loss on drying of NMT 2.0% w/w at 105°C achieved. LOD of dried granules achieved between NMT 2.0% w/w. Hence the drying process was found to comply the predefined specification for 3 batches
Pre lubrication and lubrication
The pre lubrication time of 25 min is to match number of revolution with that of tablet batches and found satisfactory at blender fast speed. The blend uniformity results were found to comply with the predefined specification. Lubrication time of 5 min at blender fast speed shows satisfactory results. Blend uniformity results found to be complied with predefined specification for all three batches. The process validation of Lacidipine tablets 6 mg was conducted for a batch size of 45.00 kg (100,000 tab) which included the validation of critical steps of manufacturing such as compression and film coating which were found satisfactory.
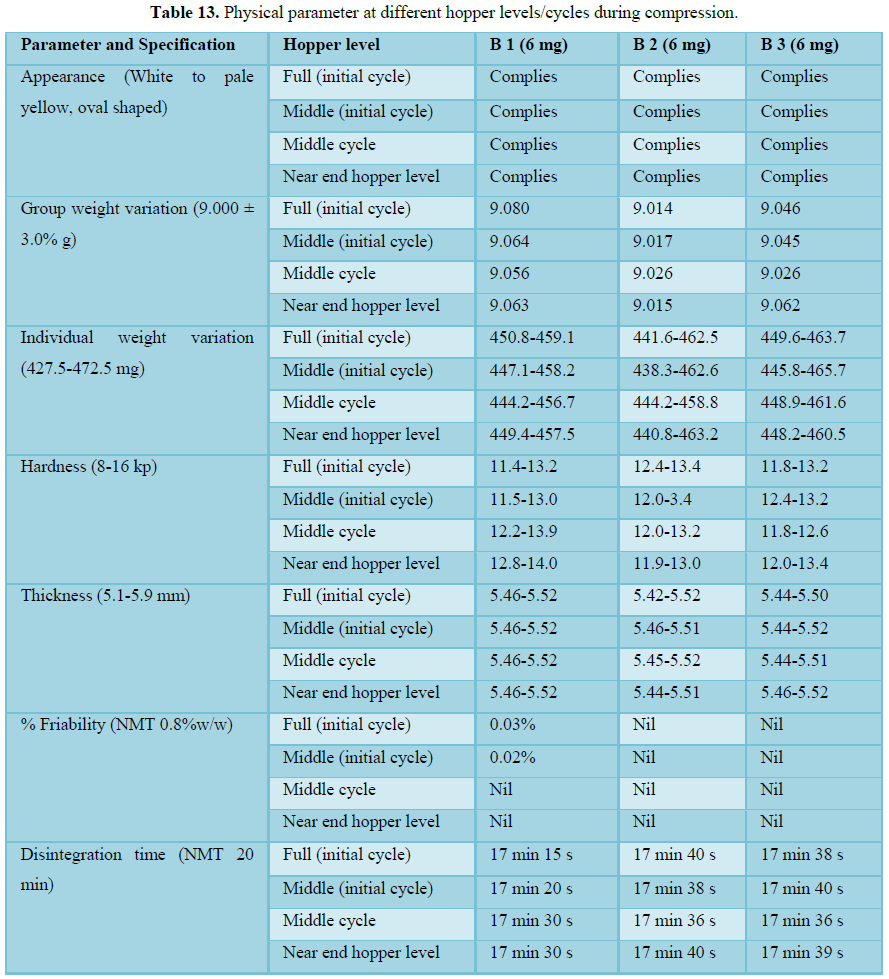
Compression
Compression was carried out on 30 station Fette press. All physical parameter such as individual weight variation, thickness, friability, disintegration time are well within the acceptance limit at full hopper, middle hopper and end hopper. Hopper study data shows no segregation during compression and uniformity of dosage unit at full hopper; middle hopper and end hopper are found satisfactory. On the basis of all analytical and physical parameter data found that compression stands validated.
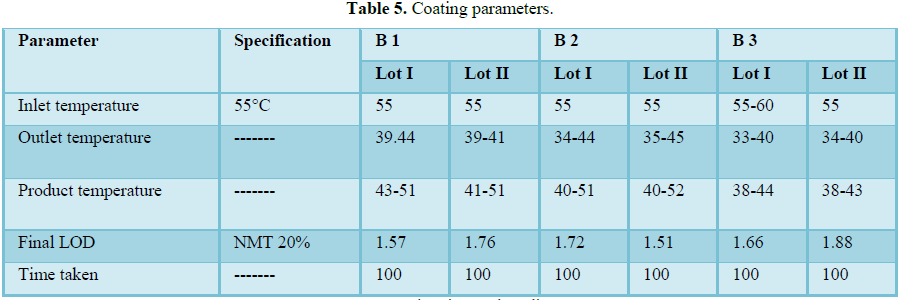
Film coating
Coating had been performed with the parameters as mentioned in manufacturing instructions in order to obtain the desired film coating buildup of 3.0 ± 0.5% w/w. Film coating inlet temperature is recommended as 65°C-75°C. Finished product report shows that final product meets the finished product specification.
Deviation and incidents: Nil.
Compression: Stands validated as per parameters specified in manufacturing instructions.
Film coating: Stands validated as per parameter specification in manufacturing instructions.
CONCLUSION
The manufacturing of three batches of common blend for Lacidipine tablets 6 mg was conducted for a batch size of 94.50 kg (210,000 tablets). The study involved validating the process variables of this transferred product to show that the process is under control. The study includes the validation of critical steps of manufacturing such as blending, drying, granulation, compression and coating. The Process validation of Lacidipine tablets showed that there was no significant batch-to-batch variation. Therefore it can be concluded that the process stands validated and the data can be used in regulatory submission for obtaining marketing authorization for the Lacidipine tablets.
1. USFDA (2004) Guideline for industry – Sterile drug product produced by aseptic process cGMP.
2. USFDA (2011) Guidelines on process validation: General principle and practice. WHO: Guidelines on GMP requirement: Part 2 - Validation.
3. TGA Guidelines (2002) Australian code of good manufacturing practice for medicinal products, pp: 103-109.
4. Beaty NA, Narlin B (1978) Aseptic vial and syringe filling. Am Chem Soc, pp: 123-128.
5. Watler P, Rathore AP, Joseph F, Edward R, Arling GS (2002) Process validation - How much to do and when to do it. BioPharm, pp: 18-28.
6. Woodcock J (2004) The concept of pharmaceutical quality. Am Pharm, pp: 1-3. (Available on: http://americanpharmaceuticalreview.com/ViewArticle.aspx ContentID
7. McBurnie L, Bardo B (2004) Validation of sterile filtration. Pharm Technol 2004: s13-s23.
8. Stockdale D (2005) Overview of aseptic fill/finish manufacturing. Part 2: Regulatory requirements. Am Pharm Rev, pp: 123-129.
9. Brett M, Belongia S (2006) Characterization, qualification and validation of disposable final filling process for parenteral and ophthalmic drug. Pharm Technol.
10. Spurgeon T (2006) Aseptic process validation is a new FDA guidance imminent? Contact Pharma. Available at: http://www.fda.gov/cber/faq/sanofiqa.htm
11. http://www.fda.gov/ICECI/EnforcementActions/WarningLetter
12. http://www.fda.gov/cber/faq/sanofiqa.htm
13. Siddiqui MS (2010) Monitoring of aseptic environments and processes in sterile facility. Available on: http://www.askaboutvalidation.com
14. Parenteral Drug Association (2011) Technical Report No. 22. Process simulation for aseptically filled products, pp: 1-40.
15. Chaurasia S, Golani S, Jain NP, Goyal M, Verma S (2011) Comprehensive review on aseptic fill/finish manufacturing as per regulatory guidelines. J Curr Pharm Res 5: 19-27.
16. Dubey SK, Basia A (2011) cGMP requirement for process control. Int J Curr Pharm Res 3: 58-63.
17. Grege G (2011) Basic requirements for aseptic manufacturing of sterile medicinal products: A comparison between Europe and USA.
18. EU Guidelines to Good Manufacturing Practice (2008) Manufacture of sterile medicinal products. Annex 1. 4: 1-16.
19. James FJ (2006) Validation of Pharmaceutical Processes. 2nd Edn. Mercel Dekker Inc., pp: 1555-1561.
20. Guidance for Industry (2004) Sterile products produced by aseptic processing current good manufacturing practices. U.S. FDA, pp: 1-63.
21. Agalloco J (2005) Importance of background microbial levels in the manufacture and testing of sterile products. Pharm Technol 74.
22. ISO 13408-1. Aseptic processing of health care products. General Requirements, pp: 1-35.
23. ISO 14644-1. Clean rooms and associated controlled environments, classification of air cleanliness.
24. Savant DA (2007) The Pharmaceutical Sciences. Pharma Pathway, 4th Edn. Pragati books Pvt. Ltd., pp: 1.91-1.99.
25. Scott B (2010) Process validation of oral solids dosage form. Part 1: General principles. Available on: http://www.ikev.org/haber/bozzonejune1.pdf
26. Syed IH (2006) Pharmaceutical Master Validation Plan. 1st Edn. St. Lucie Press, pp: 2-27.
27. White E (2009) Risk management for aseptic processing [online] pharmaceutical technology. Available from: http://www.bioline.org.br/pdf?pr02016
28. Work group of the Scottish QA Specialist Interest Group (2004) Guideline on test method for environmental monitoring for aseptic dispensing. pp: 35-40.
29. Guidance for Industry Process Validation (2008) General principle and practices. Available at: http://www.fda.gov./Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070336.pdf
30. Guidance for Industry Process Validation (1987) U.S. FDA. Available at: http://www.fda.gov./Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm124720.htm
31. Nash Robert A (2000) Pharmaceutical process validation. 3rd Edn. 129: 159-185.
-
 Table 1
Table 1 -
Table 2
-
Table 3
-
Table 4
-
Table 5
-
Table 6
-
Table 7
-
Table 8
-
Table 9
-
Table 10
-
Table 11
-
Table 12
-
Table 13
-
Table 14
-
Table 15
-
Table 16
-
Table 17
-
Table 18
-
Table 19







QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Agriculture and Forest Meteorology Research (ISSN:2642-0449)
- Journal of Womens Health and Safety Research (ISSN:2577-1388)
- Journal of Biochemistry and Molecular Medicine (ISSN:2641-6948)
- Journal of Microbiology and Microbial Infections (ISSN: 2689-7660)
- Proteomics and Bioinformatics (ISSN:2641-7561)
- Journal of Astronomy and Space Research
- Advances in Nanomedicine and Nanotechnology Research (ISSN: 2688-5476)


