Research Article
Cytotoxicity and Mutation
2852
Views & Citations1852
Likes & Shares
In this paper toxicity and gene transformation was studied. Oncogene activation and tumor suppressor gene inactivation induced by mutations provide strong evidence for the involvement of genotoxic mechanisms in tumor formation. In carcinogenesis the first stage initiation includes a genotoxic event, causing a premutagenic lesion in a single cell. In the next stage promotion finally resulting in tumor formation. The number of proto-oncogenes that must be activated to convert a normal cell into a malignant cell is unknown at present. In this paper induced toxicity and cell transformation was studied. Contribution of this paper has stopping mutation and deoxyribonucleic acid repair. Causal link between mutation and celltransformation was provided.
Keywords: DNA repair, induced toxicity, mechanism, promotion, genotoxic event, tumor formation
ABRREVIATIONS
AMP: Adenine Monophosphate; ADP: Adenine Diphosphate; AP: Adenine Phosphate; ATP: Adenine Triphosphate; DNA: Deoxyribonucleic Acid; CDP: Cytosine Diphosphate; GMP: Guanine Monophosphate; GTP: Guanine Triphosphate; RNA: Ribonucleic Acid; mRNA: Messenger RNA; rRNA: ribosomal RNA; tRNA: Transfer RNA; UV: Ultraviolet Light; UDP: Uridine Diphosphate
INTRODUCTION
Cytotoxicity resulting in cell death, is often the consequence of exposure to a harmful chemical, but the number of cells that must be killed before the function of a tissue or organism is noticeably impaired is highly variable. Some cell types, such as epithelia of the kidney and the liver, have the ability to regenerate in response to damage, while others such as neurons can not. Furthermore, some organs, such as the liver, lung, and kidney, have a substantial functional reserve capacity in excess of normal requirements, and normal function can be maintained even in the presence of extended necrosis [1-5].
In additional to cell death, disturbances in the regulation of cell division induced by toxic xenobiotics may have harmful long-term consequences for the organism affected. Nonlethal alternations in the genome of somatic cells have been known to result in mutations and possibly lead to malignant transformation and tumor formation [6-12]. During the last years, evidence has shown that compounds not directly interacting with the genomic DNA can also produce cancer by so called epigenetic mechanism. These may involve a proliferative response of epithelial cells to cytotoxicity, which has been suggested to occur with high dose carcinogens chemicals [13-21]. In this paper induced toxicity to genes transformation was studied.
DNA function
The nucleotides participate in a wide variety of biochemical processes. Perhaps the best-known role of purine and pyrmidine nucleotides is to serve as the monomeric precursors of RNA and DNA [22-26]. However, the purine ribonucleotides serve also as the ubiquitous high energy source, ATP, as regulatory signals (cycle AMP [cAMP] and GMP [cGMP]), and as components of the coenzymes and of the methyl group donor S adenosil methionine. The pyrimidine nucleotides in addition to providing monomeric precursors for nucleic acid synthesis, also serve as high energy intermediates, such as UDP-glucose and UDP-galactose in carbohydrate metabolism and CDP-acylglycerol in lipid synthesis.
The heterocyclic bases purine and pyrimidine are the parent molecules of nucleosides and nucleotides. Nucleotides are ubiquitous in living cells, where they perform numerous key functions. Examples include incorporation, as their ribose (RNA) or deoxyribose (DNA) monophosfates, into nucleic acids, energy transduction (ATP), parts of coenzymes (AMP) acceptors for oxidative phosphorylation (ADP) allosteric regulators of enzyme activity, and second messengers (cAMP), (cGMP).
The heterocyclic bases purine and pyrimidine are the parent molecules of nucleosides and nucleotides. Nucleotides are ubiquitous in living cells, where they perform numerous key functions. Examples include incorporation, as their ribose (RNA) or deoxyribose (DNA) monophosfates, into nucleic acids, energy transduction (ATP), parts of coenzymes (AMP) acceptors for oxidative phosphorylation (ADP) allosteric regulators of enzyme activity, and second messengers (cAMP), (cGMP).
Biomedical important it neither nucleotides nor their parent purine and pyrimidine bases in the diet are incorporated into human tissue nucleic acids or into purine or pyrimidine coenzymes. Even when a diet rich in nucleoproteins is ingested, human subjects form the constituents of tissue nucleic acids from amphibolic intermediates. This de novo synthesis permits purine and pyrimidine analogs with potential as anticancer drugs to be incorporated into DNA. The rates of synthesis of purine and pyrimidine oxy- and deoxyribonucleotides are subject to precise regulation. Mechanisms have evolved to ensure production of these compounds in quantities and at times appropriate to meet varying physiologic demand.
In addition to de novo synthesis, these include “salvage” pathways for reutilization of purine or pyrimidine bases released by degradation of nucleic acids in vivo. Human diseases that involve abnormalities in purine or pyrimidine metabolism include gout, Lesch-Nyhan syndrome, Reye’s syndrome, adenosine deaminase deficiency, and purine nucleoside phosphorylase deficiency.
Induced toxicity
The linkage between in the nucleus and proteins in the cytoplasm is not direct. Information contained in DNA molecule is transferred to the protein synthesizing machinery of the cell via messenger RNA (mRNA), which is synthesized complementary to the relevant DNA sequences by RNA polymeraze. The mRNA molecules act as transport vehicles for information contained in the genes being expressed (Silva- Oliveria; Silva, Martinho, Cruvinel-Carloni, 2016 [22-26].
In eukaryotic cells, the initial mRNA copy contains homologues of both the intron and the exon regions. The intron region are removed and the exon regions are spliced together to form the active mRNA molecules, which are then transported through the pores of the nuclear membrane to the cytoplasm.
The next process involves the translation of mRNA molecules into polypeptides. This procedure requires many enzymes and two further types of RNA: transfer RNA (tRNA) and ribosomal RNA (rRNA). A specific tRNA exists for each amino acid. The tRNA molecules are involved in the transport anticodon of an appropriate incoming tRNA –amino acid complex and coupling of amino acids into the resulting polypeptide. Each tRNA molecule has two binding sites: one for the specific amino acid, the other containing a tripler of bases (the anticodon) that is complementary to the appropriate incoming mRNA.
The rRNA is complexed with protein to form subcellular globular organelles called ribosomes. Ribosomes can be regarded as the reading heads, which allow the linear array of mNRA codons to base pair with an All cells possess the same genetic information; however, different types of cells exhibit distinct gene transcription patterns. These differences in gene expression are critical to the morphological and biochemical properties of the many thousands of cell types in the human and animal body. Hence, mechanisms are required that regulate gene expression, determine which genes are expressed and to what extent and which genes are not expressed in a certain cell type at a particular time. The mechanisms involved in regulation of gene transcription are not entirely understood. The transcription of structural genes is regulated by a special set of codons, in particular, promoter sequences, the initial binding sites for RNA polymerase before transcription begins. Different promoter sequences have different affinities for RNA polymerases. Additional regulatory genes called operators regulate the activity of several genes or gene groups (operons).
The activity of the operator itself is further controlled by a repressor protein, which stops transcription of the whole operon by binding to the operator sequence. Because of these regulatory mechanisms, cells are able to express only the genes required at a given moment for their specialized function. This not only helps to converse cellular energy but also is decisive for correct cellular differentiation, tissue pattern formation and function, and maintenance of the physiological integrity of the entire organism.
All living cells possess several efficient DNA repair processes. All living cells possess several efficient repair processes. Repair is crucial in protecting cells from spontaneous and exogenous lethal and mutating effects such as heat–induced hydrolysis, UV light, ionizing radiation, DNA-reactive chemicals, free radicals, and reactive oxygen species. Among the various existing repair mechanisms, the most comprehensively studied mechanism in eukaryotes is the excision repair pathway. The mechanism includes a group of enzymes acting cooperatively to recognize . Lesions, remove them, and correctly replace the damaged sections of [10,16].
The excision repair pathway is regarded as error free and does not lead to mutations. However, this pathway may become saturated after excessive damage. In this case, the cell may be forced to activate other repair mechanisms that are not error free. Several of these mechanisms, such as error-prone repair, have been well characterized in bacteria, but their counterparts, if any, in mammalian cells have not been identified.
Mutations
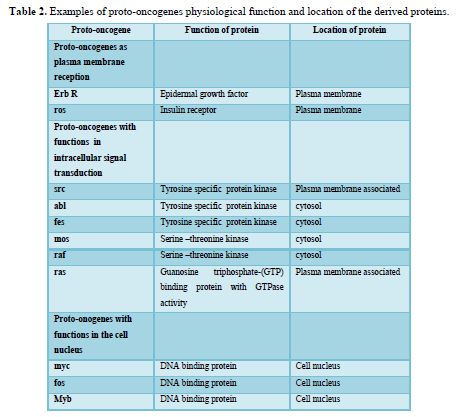
Mutations are hereditary changes in genetic information, resulting from spontaneous or chemicals induced DNA damage [19]. The term mutation can be applied to point mutations, which are qualitative changes involving one or a few bases within one gene, as well as to larger changes involving parts of the chromosome detectable by light microscopy or even whole chromosomes and thus many thousands of genes as shown in (Table 1).
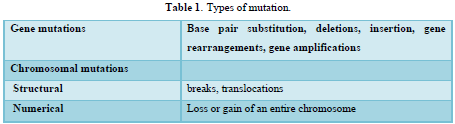
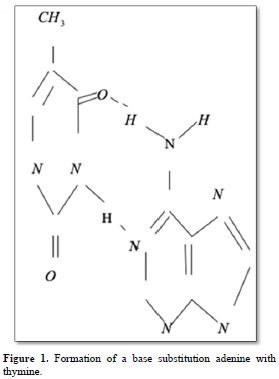
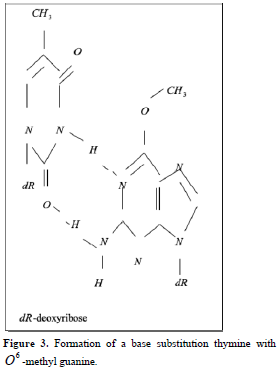
Point mutations can occur when one base is substituted for another, base substitutions, or when base pairs are deleted or inserted, deletions/insertions. Substitution of another purine base or of another pyrimidine for a pyrimidine base is called transitions, while substitution of purine for pyrimidine or pyrimidine for purine is called transversion. Very slight alterations in the chemical structure of the DNA bases may be sufficient for a base pair substitution to occur. Guanine for example, normally pairs with cytosine, while -methyl guanine (a frequent DNA modification seen with ethylating agents such as dimethylnitrosamine) pairs with thymine as sohwn in Figures 1, 2 and 3. These changes in certain codons may cause insertion of the wrong amino acid into a relevant polypeptide. In this case, the changes are called missense mutations. Such proteins may have dramatically altered properties if the new amino acid is close to the active center of an enzyme or affects the three-dimensional structure of an enzyme or a structural protein. Hence, the alterations may result in marked changes in the differentiation and proliferative characteristics of the affected cells. A base substitution can also result in the formation of a new inappropriate stop or nonsense codon. The result of nonsense mutations is the formation of a shorter and, most likely, inactive protein.
Owing to the redundancy of the genetic code, about a quarter of all possible base substitutions will not result in amino acid replacement and will be silent mutations. Bases can be also deleted or added to a gene.
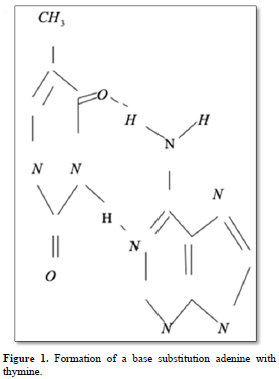
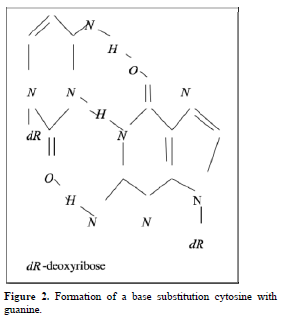
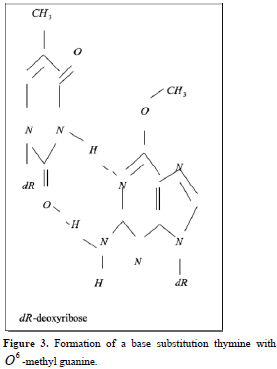
Because each gene has a precisely defined length, these changes, if they involve a number of bases that is not a multiple of three, result in a change in the reading frame of DNA sequence and are known as frame shift mutations. Such mutations often have a dramatic effect on the polypeptide coded by the affected gene, because most will differ from the point of the insertion or deletion of bases in the DNA strand onward.
Some forms of unrepaired alkylated bases are lethal, due to interference with DNA replication. Others such as -methyl guanine lead to mutations, if unrepaired. These differences indicate that not all DNA adducts are of equivalent importance. In fact, some adducts appear not to interfere with normal DNA functions or to be rapidly repaired, others are mutagenic, and yet others are lethal. The most vulnerable base is guanine, which can form adducts at several of its atoms, e.g. N-7, C-8, O-6, and exocyclic N-2.
Cross-links
Chemicals with bifunctional alkylating properties can also form links between adjacent bases on the same strand (intrastrand cross-links) or crosslinks between bases on different strands (interstrand).
The induction of a frame shift mutation does not necessarily require formation of covalent adducts. Some compounds with a planar structure, such as particular polyclyclic aromatic hydrocarbons, can intercalate between strands of the DNA double strand. The intercalated molecules my interfere with DNA repair or replication and cause insertions and/or deletions of base pairs.
The precise molecular event is still unclear, although several mechanisms have been proposed. Hotpots for frame shift mutations often involve section of the DNA strand where there is a run of the same base (e.g. the addition of a guanine to a run of six guanine residues).
DNA strand breaks result from hydrolysis of the sugar –phosphate bond of nucleotide. In a double strand, both single– and double-strand breaks may occur. DNA strand breaks strand breaks are often induced by hydroxyl radicals, which are formed at high rates both spontaneously during normal cell life and to the presence of exogenous chemicals. AP lesions in the DNA strand result from spontaneous hydrolysis of the glucosidic bond and loss of the DNA base. Similar to DNA strand breaks, AP lesion are common spontaneous events, however, hydrolysis can be increased dramatically by various types of DNA adducts, such as the substitution of purines, a common target of alkylating chemicals. Another common spontaneous event is the deamination of cytosine to uracil, approximately 100 deaminations take place in each cell every day.
Causal link between mutation and cancer
The change from cells undergoing normal controlled cell division and differentiation to cells that are transformed, dividing without control, and undifferentiated or abnormally differentiated does not occur as a single step. Malignant transformation is a multistage process. Evidence for the involvement of multistage comes from in vitro studies, animals models, and epidemiological observations. In humans, the latent period between exposure to a chemical carcinogen and the operations of a tumor in the target tissue is approximately 10-25 years.
Modern molecular biology techniques enable thorough investigation of the genome of malignant cells compared to the genome of their normal counter parts. These studies clearly show that a single mutation is not sufficient to induce malignant transformation. The number o genetic changes vary between two and seven in different tumor types. Also, several types of mutations are usually formed in a malignant transformed cell, e.g. base pair substitutions, gene rearrgements, chromosomal breaks, and deletions [6,7].
Proto-oncogenes and tumor suppressor genes
Normal control of cell division and differentiation is now generally accepted to based on the interplay of two sets of genes, the proto-oncogenes and the tumor suppressor genes. Abnormal activation of proto-onogenes or inactivation of tumor suppressor genes eventually leads to malignant transformation [9,18].
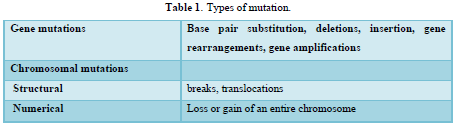
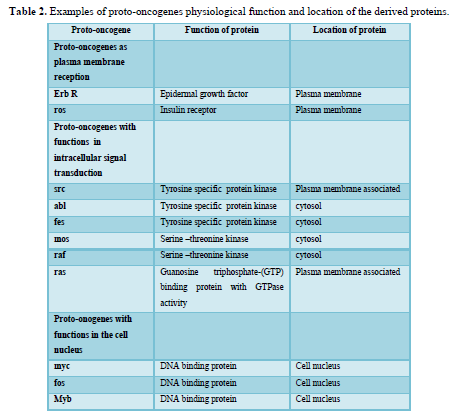
Oncogenes were originally discovered in the genome of transforming retroviruses and were therefore named v-oncogenes. Subsequent studies showed that these viral genes were originally derived from the mammalian genome. In the normal cell, these proto-oncogenes have important functions in signal transduction pathways (Table 2). Proto-oncogenes are expressed in the course of physiological growth processes, such as organ formation during embryogenesis in the maternal organism, regeneration of damaged tissues, and stimulation of cell division by growth factors in the adolescent and adult organism. Activation of cellular proto-oncogenes into c-oncogenes in spontaneously and chemically induced tumors results in qualitatively (altered protein) or quantitatively, too much, at the wrong time altered gene expression. Oncogenes can be activated by several types of genetic damage such as point mutations, gene amplifications, and chromosomal translocations.
A substantial number of human tumors 10-15% contains activated ras oncogenes. In all cases so far examined, activation relies on a point mutation in codon 12, 13, 59, or 61 of one of the ras proto-oncogenes. The p21 protein coded by the ras genes exerts GTPase (GTP hydrolase) activity and is membrane bound. The physiological function of p21 is not precisely known; the protein is bound to the inner surface of the plasma membrane; therefore, it may have a function in transducing signals from growth factors receptors across the cytoplasm to the nucleus. The point mutations described above change the properties of the p21 protein and often result in reduced GTPase activity. Since this activity is also responsible for inactivation of the p21 protein itself, reduction of GTPase activity by point mutation results in prolongation of the ras signal transduction pathway.
Point mutations resulting in activation of ras proto-oncogenes have also been described in several chemically induced rodent tumors. In the majority of mammary tumors induced by nitro-somethylurea, the ras oncogene is mutated at codon 12. Similar activation of H-ras at codon 61 is detected in mammary and skin tumors induced by 7, 12- dimethylbenzanthracene. Nitro-somethylurea is an alkylating agent that induces the formation of -methylguanine in DNA which is consistent with the type of point mutation (G transition) observed at the 12th codon of H-ras. The mutation at codon 61 in dimethylbenzanthracene-induced tumor cells (mainly A T transversion) is consistent with the formation of adenine adducts, resulting from dimethylbenzanthracene metabolites binding to adenine residues.
CONCLUSION
This paper considers toxicity and malignant transformation and intrastrand and interstrand cross links. Causal link between mutation and cancer was studied. Tumor suppressor genes code for proteins that negatively regulate cell proliferation and inhibit neoplastic transformation. Oncogenes act in a dominant manner, i.e. activation of one copy of the gene can result in perturbation of normal cell proliferation or diferentation.
- Stevanovic JS, Djurovic J (2012) Pharamceutical drugs formation modelling, 4th International Conference on Drug Discovery and Therapy, Dubai, UAE, 12-15.
- Stevanovic JS (2012) Helix stability of the double to single strands oligonucleotides transition, HSBS’12- Health Science and Biomedical Systems, Naun, Romania, Iashi, June 13-15.
- Ihara Chem Ind US 4289916 (1980) (Y.Nakayama).
- Ihara Chem Ind JP 60125251 (1983) (J.Kiji).
- Stevanovic JS, Grkinic LJV, Marinkovic SS (2013) Molecular modeling of the helix structure formation in biopolymers, WCCE9-9th World Congress of Chemical Engineering, P-01-136, Coex, Seoul, Korea, August 18-23.
- Harris CC, Weston A, Willey JC, Trivers GE, Mann DL (1987) Biochemical and Molecular Epidemiology of Human Cancer: Indicators of Carcinogen Exposure, DNA Damage, and Genetic Predisposition. Environ Health Perspect 75: 109-119.
- Lawley P (1989) Mutat Res 213: 3-26.
- Bishop ЈМ (1987) Science (Washington DC 1883) 235: 305-311.
- Stevanovic JS (2010) Nucleotides recombination, states. Trasact Biol Biomed 7(3): 253-262.
- Stevanovic JS (2010) DNA combination and recombination state, 10the International Conference on Mathematics and Computers in Biology and Chemistry, Iashi, pp: 11-12.
- Guengerich FP (1991). J Biol Chem 266: 10019-10022.
- Cmarik JL (1990). Cancer Res 50: 2747-2752.
- Ziegler DM (1988). Drug Metab Rev 19: p1-32.
- Hines RN (1994). Toxicol Appl Pharmacol125: 1-6.
- .Stclair GMB (1988). Chem Res Toxicol 1: 179-185.
- Bock KW, Lipp HP, Block-Hening BS, BS (1990) Xenobiotica 20: 1101-1111.
- Blum M (1990). DNA Cell Biol 9: 193-203.
- Bishop JM (1991). Cell 64: 235-248.
- Stevanovic JS (2018) Toxic effects and DNA transformation. Biochem Biotechnol Res 6(2): 15-19.
- Stevanovic JS. (2020) Mutation by base pair substitution. Adv Nanomed Nanotechnol Res 2(2): 122-125.
- Stevanovic JS (2020) Using recombinant DNA technology against viruses and bacterias. J Genet Eng Biotechnol Res 2(2): 1-6.
- Silva-Oliveria RJ, Silva VAO, Martinho and Cruvinel-Carloni A (2016) Cytotoxicity of allitinib, an irreversible anti EGFR agent, in a large panel of human cancer-derived cell lines: KRAS mutation status as a predictive biomarker. Cellular Oncol 39 (3): 253-263.
- House IG, Thia K, Bernnan AJ (2015) Tothil, Immunology and cell, John Wiley& Sons Online Library.
- Granja S, Pinheiro C, Manuel Reis R, Martinho O, Baltazar (2015) Glucose addiction in cancer therapy: Advances and drawbacks. Curr Drug Metab 16(3): 221-242.
- Buglewicz DJ, Mussallem JT, Haskins A H, Su C, Maeda J, et al. (2020) Cytotoxicity and Mutagenicity of Narrowband UVB to mammalian cells. Genes 11: 646.
- Smith CJ, Perfetti (2020) Statistical treatment of cytotoxicity in Ames bacterial reverse mutation assays can provide additional structure activity relationship information. Toxicol Res Appl.
-
 Table 1
Table 1 -
Table 2

QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Womens Health and Safety Research (ISSN:2577-1388)
- Journal of Genetics and Cell Biology (ISSN:2639-3360)
- Journal of Astronomy and Space Research
- Journal of Microbiology and Microbial Infections (ISSN: 2689-7660)
- Advances in Nanomedicine and Nanotechnology Research (ISSN: 2688-5476)
- Journal of Biochemistry and Molecular Medicine (ISSN:2641-6948)
- Journal of Veterinary and Marine Sciences (ISSN: 2689-7830)


