| Mariko Witalis#, Philippe St-Onge# and Woong-Kyung Suh* |
| Corresponding Author: Woong-Kyung Suh, 110 avenue des Pins Ouest, IRCM, Montreal, QC, H2W 1R7, Canada, Tel: 514-987-5720; Fax: 514-987-5768; E-mail: woong-kyung.suh@ircm.qc.ca |
| Received: September 1, 2015; Revised: April 28, 2016; Accepted: November 12, 2015 |
| DOI: #Authors contributed equally to this work |
| Citation: Witalis M, St-Onge P, Suh W. -K.(2016) Angioimmunoblastic T Cell Lymphoma (AITL): Origin, Pathogenesis and Therapeutic Options. J Immunol Res Ther, 1(1): 29-36. |
| Copyrights: ©2016 Witalis M, St-Onge P, Suh W. -K. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| Share : |
1306
Views & Citations306
Likes & Shares
INTRODUCTION
Angioimmunoblastic
T cell lymphoma (AITL) is a late-onset
peripheral T cell lymphoma, representing 18.5% of mature T/NK cell malignancies
[1]. Symptoms include generalized lymphadenopathy, hypergammaglobulinemia, and
hemolytic anemia with poor prognosis (five-year overall survival ~33%). AITL
tumors display effacement of the lymph node (LN) architecture with prominent
arborization of endothelial venules. Because this malignancy manifests in
elderly patients (avg onset ~64 yr), aggressive interventions are typically
avoided and conventional chemotherapy has been the sole treatment. However,
multiple variations of chemotherapy regiments of AITL have shown poor efficacy
and more innovative treatment is eagerly awaited [2]. Importantly, an
accumulating body of evidence indicates that AITL originates from T follicular
helper (Tfh) cells, probably associated with germinal center (GC) reaction. In
this review, we will summarize data that support the Tfh origin of AITL and
propose a model that may explain the etiology of AITL. At the end, we will
discuss T cell costimulatory signaling pathways as potential therapeutic
targets for AITL.
AITL arises
from Tfh
Tfh cells are a
subset of CD4 T cells whose differentiation is driven by the lineage-specifying
factor Bcl6 [3]. Tfh cells express CXCR5 chemokine receptor, inducible T cell
costimulatory receptor (ICOS), and programmed cell death protein 1 (PD-1) on
the cell surface and produce IL-4 and IL-21 as effector cytokines.
Tfh cells have
the unique ability to migrate into B cell follicles in secondary lymphoid
organs to form GCs. The GC is a microenvironment in which antigen-specific B
cells are selected through interaction with Tfh cells to become either antibody
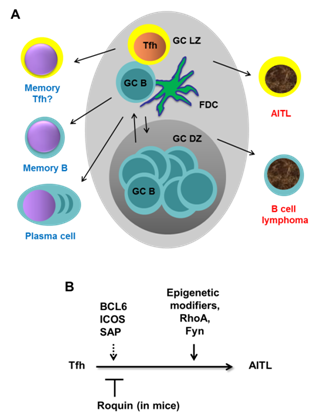
producing plasma cells or memory B cells [4] (Figure 1A). Since failure to
produce Tfh cells leads to immunodeficiency whereas excessive Tfh responses
induce autoantibodies, the generation and function of Tfh cells is highly
regulated by multiple molecular and cellular mechanisms [5,6]. Precursor Tfh
(Pre-Tfh) cells are generated during dendritic cell-mediated T cell priming
under the influence of IL-6 and IL-21 (mouse) or IL-12 (human). However, the
pre-Tfh population fails to fully differentiate into mature GC Tfh cells unless
they interact with cognate B cells to reinforce their differentiation programs
by upregulating Bcl-6. The intimate T-B interactions during GC reactions depend
on T cell costimulatory signals mediated by multiple T cell costimulatory
receptors (see below).
Multiple lines
of evidence strongly suggest that Tfh cells are the origin of
AITL [7-15]. First, although T cells represent a small fraction of the total
tumor mass, it was confirmed that T cells are the ones that undergo neoplastic
transformation [12]. This expansion of Tfh-like tumor cells appears to cause
expansion of B cells and infiltration of other immune cells. Secondly,
immunohistochemical studies have shown that AITL tumors express high levels of
Tfh markers: PD-1, CXCL13, ICOS, SAP, and BCL6 [9-11,13]. Thirdly, gene
expression profiling of highly enriched T cells from AITL samples revealed “Tfh
signatures” [7,8,14]. Lastly, in a mouse model of AITL termed Roquinsan
+/-mice (see below), abrogation of genes that are known to be crucial
for generation of Tfh cells and germinal center formation (such as CD28, ICOS
and SAP) dramatically reduced AITL-like tumor incidence [15]. Together, these
findings strongly support the idea that AITL arises from Tfh cells during
germinal center reaction.
How does AITL
arise?
Lessons from
human cancer genomics
Although it is
becoming clear that AITL arises from Tfh cells, the genetic alterations causing
the transformation remain poorly understood. Recently, three studies using
genome-wide sequence analysis shed light on this issue [16-18]. These studies
unanimously revealed high frequency of recurrent point mutations in the small
GTPase RHOA gene and multiple genes involved in epigenetic
modification and T cell signaling.
The Gly17Val (G17V) mutation in RHOA gene is present in
53-68% of AITL tumor samples [16-18]. This mutation has been uniquely found in
AITL but not in other types of T cell, B cell, or myeloid malignancies [17,18].
Mechanistically, Gly17Val mutation abrogates the GTP binding ability of RHOA
[16] increasing its association with guanine-exchange factors [16,17]. This may
explain why G17V RHOA protein shows dominant negative functions in
transcriptional regulation and actin remodeling when overexpressed. One study
has shown that, when overexpressed in Jurkat leukemia cells, G17V RHOA enhanced
cellular proliferation and invasiveness [18]. However, similar experiments by
another group showed no differences in proliferation [17]. Therefore, it
remains to be seen how and to what extent this dominant negative mutation of RHOA contributes
to the transformation process of AITL.
A group of
mutations frequently found in AITL belong to genes encoding epigenetic
modifiers: Tet methylcytosinedioxygenase 2 (TET2), DNA
(cytosine-5)-methyltransferase 3A (DNMT3A), and isocitrate dehydrogenase 2
(IDH2) [16-18]. TET2, DNMT3A, and IDH2 dynamically modulate methylation status
of DNA and histones directly or indirectly affecting gene expression profiles.
These genes are altered in multiple hematologic malignancies including AITL
[19-21]. Importantly, some mutations in TET2 and DNMT3A genes
were discovered in both non-tumor and tumor hematopoietic cells [17].
Interestingly, in this study, all the RHOA mutated AITL tumor
samples also had TET2 mutations. This suggests that TET2 mutation
precedes and possibly be a prerequisite of RHOA mutation
during a multi-step process of AITL transformation [17]. However, Palomero et
al. documented that approximately 25% of AITL tumors have RHOA mutation
without TET2 mutation arguing against the sequential mutation
events [16].
In addition,
Palomero et al. identified a less prevalent (3% of AITL), yet
mechanistically well-defined mutation in the FYN gene. FYN is
a member of SRC family kinase that plays an important role in TCR signaling
[22]. Three different mutations were found in FYN: two in the SH2
domain and one in the C-terminal inhibitory tyrosine residue. These mutant FYN
proteins showed higher kinase activity, most likely because these residues are
responsible for keeping FYN in an inactive conformation. The authors also showed
that the activity of mutant FYN could be inhibited by a SRC kinase inhibitor
dasatinib suggesting that this subset of AITL may be treated by dasatinib.
Overall, the
data suggest that AITL arises through the accumulation of multiple somatic
mutations in genes involved in epigenetic modifications, small GTPase RHOA, and
TCR signaling components. However, several questions remain to be answered
regarding the role of these mutations in the pathogenesis of AITL. First, what in
vivo roles does each individual mutation play? Second, how do these
multiple mutations cooperate to culminate in cellular transformation? Third, is
there a “driver” mutation that initiates a cascade of events leading to AITL or
is the accumulation of independently arising multiple mutations sufficient?
Fourth, what are the cell-intrinsic and/or extrinsic factors that promote the
accumulation of the AITL signature mutations in Tfh cells in the GC? Many of
the questions listed above could be addressed through the use of genetically
modified mouse models.
An AITL mouse
model
Recently,
Vinuesa and colleagues found that mice possessing one copy of the san allele,
a dominant negative point mutation of the Roquin gene (Roquinsan/+ mice),
develop an AITL-like disease: asymmetric LN tumors, oligoclonal expansion of
Tfh cells, effacement of the LN architecture, arborization of endothelial
venules, and hypergammaglobulinemia [15]. The san point
mutation of Roquin is known to dysregulate the stability of
numerous mRNA species including that of ICOS in CD4 T cells due to the
disruption of Roquin-mediated mRNA degradation [23,24]. This leads to
lupus-like autoimmune disease admixed with AITL-like disease in all Roquinsan/san mice
within 4-months of age [25]. In contrast, Roquinsan/+ mice
do not develop lupus-like disease but still manifest AITL-like disease (~50%
penetrance at 6-months of age) [15]. This work prompted a study with a cohort
of AITL patients to see if alterations of ROQUIN expression levels or mutations
are associated with AITL [26]. No alterations in the ROQUIN locus
or its mRNA and protein expression levels were detected in this study
suggesting that alteration of ROQUIN does not normally happen during AITL
development. One possibility is that Roquin heterozygosity may
represent one of many mechanisms that promote hyperactive Tfh program which
leads to mutation of AITL-causing genes. However, further work is required to
establish a potential relationship between Roquinsan mutation
and more direct oncogenic events that cause AITL-like disease in this mouse
model. Meanwhile, Roquinsan/+ mice may offer
opportunities to study molecular pathogenesis and discover therapeutic targets
for AITL. For example, consistent with the notion that AITL originates from Tfh
cells within the GC, germline ablation of genes that are critical for Tfh
generation and germinal center formation, such as ICOS and SAP, drastically
reduced the incidence of tumors in Roquinsan/+mice [15].
Another important question that can be addressed using this mouse model is the
contribution of multiple T cell costimulatory receptors and their downstream
signaling pathways in the disease progression of AITL (see below).
What drives
AITL pathogenesis?
It is well
established that several B cell lymphomas such as Burkitt lymphoma, follicular
lymphoma, and diffuse large-cell B cell lymphoma (DLBCL) are derived from GC B
cells [27]. As discussed above, new pieces of evidence indicate that Tfh cells
are also at risk of becoming lymphomas. What could be the driving force for
transformation of these GC-derived B and T lymphomas? For B cell lymphomas, it
can be readily explained by the biology of GC B cells. Selection of
class-switched, affinity matured B cell clones involves DNA remodeling and
proliferation at the GC B cell stage. Failure of this delicate coordination can
lead to a rapid expansion of B cells that carry oncogenic legions, which are
typically chromosomal translocations that allow dysregulated expression of
oncogenes such as c-MYC (Burkitt lymphoma), BCL2 (follicular lymphoma), or BCL6
(DLBCL). It remains elusive how Tfh cells are driven to become cancer. In
contrast to GC B cells, Tfh cells do not undergo DNA rearrangement within the
GC. This is reflected in findings that somatic mutations identified in AITL
cells are predominantly point mutations but not chromosomal translocations
[16-18].
A common
feature of GC B cells and Tfh cells is the high level of BCL6 expression. BCL6
is absolutely required for the regulation of genes involved in Tfh or GC B cell
differentiation, but can also act as an oncogene when its expression is
deregulated [3,28]. Dysregulated expression of Bcl6 in mice modeled after a
human DLBCL legion led to progressive lymphoproliferation and DLBCL-like
disease [29]. In another mouse model, constitutive Bcl6 expression in T and B
cells led to low level T and B lymphomas [30-32]. However, when these mice were
exposed to other oncogenic hits the incidence of T cell lymphoma increased
dramatically. In B cells, the oncogenic effects of BCL6 is likely mediated by
its ability to enhance proliferation by inhibiting cell cycle control
mechanisms [33] and rendering GC B cells more tolerant to DNA damage by
suppressing DNA damage response pathways [34]. Whether BCL6 plays similar roles
in T cells remain to be tested. However, there is evidence that Bcl6-positive
Tfh cells are more proliferative than their Bcl6-negative counterparts in mice
[35]. Consistent with this, when single cell
suspensions of human AITL tumors were serially passaged in immune-deficient
mice, BCL6-positive, putative neoplastic AITL cells outcompeted BCL6-low T
cells and B cells [13]. Although it is yet to be confirmed in humans, a study
in mice showed that Tfh cells normally downregulate BCL6 after GC reaction and
concomitantly lose proliferative capacity [36]. Taken together, these results
suggest that AITL cells maintain a high level of BCL6 expression and that this
might be necessary for their neoplastic behavior.
Collectively,
these data support the idea that a high level of BCL6 in GC B cells and Tfh
cells
support
oncogenic processes. However, more work is required to establish how and to
what extent Bcl6 promotes AITL transformation.
Can we stop
AITL progression?
Once
transformed, AITL cells grow and spread to other lymph nodes [12].
Understanding molecular pathways that are involved in this disease progression
should help design better therapeutic options. In this regard, what we know
about the role of the T cell costimulatory receptors ICOS and SLAM family receptors
may help improve treatment of AITL.
ICOS is an Ig
superfamily transmembrane receptor belonging to the CD28 family [37]. ICOS is
expressed in T cells after activation and binds to ICOS ligand (ICOSL)
expressed on the surface of B cells and other cells [38]. In contrast to CD28
which regulates T cell expansion through induction of IL-2, ICOS mainly
regulates effector cytokines (IL-4 and IL-21) and cellular mobility [39-41].
One of the major roles of ICOS is to regulate Tfh generation and function,
hence ICOS- or ICOSL-deficiency leads to primary immunodeficiency both in
humans [42-44] and mice [45-49]. On the other hand, dysregulated ICOS/ICOSL
expression and function are highly associated with antibody-mediated autoimmune
diseases in humans (e.g., rheumatoid arthritis) [50,51] and mice (e.g., Roquinsan/san mice)
[23,25]. We and others have shown that ICOS can induce two intracellular
signals: phosphoinositide 3-kinase (PI3K) and intracellular calcium flux
[39,40]. By generating a knock-in mouse line termed ICOS-Y181F in which
ICOS-PI3K signaling is selectively abrogated, we demonstrated that the
ICOS-PI3K signaling axis promotes formation of Tfh cells in part through the
transcriptional control of key Tfh cytokines IL-4 and IL-21 [39]. Importantly,
IL-4 and IL-21 promote B cell growth and differentiation and IL-21 can serve as
an autocrine growth factor for Tfh cells [52,53]. Further, we uncovered
ICOS-PI3K-mTOR signaling in CD4 T cells that has a capacity to acutely increase
the translational efficiency of IL-4 during antigen-specific T-B interactions
[54]. A more recent in vivo imaging study indicates that
ICOS-PI3K signaling plays a critical role in keeping pre-Tfh cells motile in
the T-B border to enhance the chance for pre-Tfh cells to encounter cognate B
cells [41], a prerequisite for stable T-B conjugate formation and entry into
the GC. Lastly, ICOS signaling has an ability to enhance the survival of
activated CD4 T cells in settings of cancer immunotherapy [55,56] and
autoimmune disease [57]. Apart from PI3K, we have shown that ICOS can also
induce intracellular calcium flux in a PI3K-independent manner [39]. This
ICOS-calcium signaling promotes a feed-forward loop between ICOS and the CD40
pathway, enhancing cytokine production during T-B collaboration within the GC
[58,59]. Therefore, ICOS utilizes PI3K and calcium-mediated signaling pathways
to promote T cell motility, cytokine production, survival, and T-B crosstalk.
It is conceivable that these signaling components may continue to operate even
after Tfh cells have become neoplastic.
Another key
protein that is critical for the maturation and function of Tfh cells is the “signaling lymphocytic activation molecule
(SLAM)-associated protein” (SAP), an adaptor
protein for SLAM family receptors [60-62]. SLAM family receptors (nine members
so far identified) are widely expressed in hematopoietic cells. Mostly through
homotypic interactions, SLAM receptors mediate antibody responses,
cytotoxicity, adhesion, autoimmunity, and lymphocyte development [61,62]. Importantly,
defective SAP functions cause X-linked lymphoproliferative disease (XLP) in
human patients [63]. XLP is characterized by abnormal responses to Epstein-Barr
virus infections, lymphoproliferative syndromes, and immunoglobulin
deficiencies caused by defective Tfh cells. SAP has a single SH2 domain that
binds to immunotyrosine switch motifs located in the cytoplasmic tails of
several SLAM family receptors including CD84, Ly108, and SLAM. Studies using
knockout and knock-in mouse lines delineated Fyn-dependent or independent SAP
signaling. SAP can recruit Fyn to ligated SLAM family receptors to augment IL-4
production in CD4 T cells [64]. However, SAP can promote GC reactions and
antibody responses independently of the SAP-Fyn signaling axis [65-67]. Fyn-independent
SAP signaling plays a critical role in the formation of stable T-B conjugates
in the mid-phase of GC reaction, a prerequisite for the formation of functional
Bcl-6hi GC Tfh populations [68-70]. The SLAM family members
that are mainly responsible for this appear to be CD84 and Ly108 [69]. Although
SAP might also be expressed in B cells, it is clear that SAP expression in T
cells is both sufficient and necessary for GC reaction [65-67]. In summary,
SLAM family receptors such as CD84 and Ly108 enhance T-B adhesion and cytokine
production through SAP-mediated signaling in T cells at mid- and late-phase of
GC reaction.
Since ICOS and
SAP promote Tfh cell motility and T-B collaboration, they may play a critical
role in the progression of Tfh-derived neoplastic AITL tumor cells. Consistent
with this idea, ICOS and SAP are highly expressed in T cells from human AITL
samples [10,11]. Therefore, one could predict that interruption of T-B
interactions through blockade of ICOS or SAP signaling may slow or stop tumor
cell expansion and dissemination.
CONCLUSION
AITL is the second most prevalent mature T/NK malignancies for which
better therapeutic options are eagerly sought [2]. Pathological observations,
gene expression profiling, and cancer genome sequencing studies strongly
suggest that AITL arises from Tfh cells through accumulation of multiple
somatic mutations (Figure 1). We propose that sustained BCL6 expression and
hyperproliferation, common features of Tfh and GC B cells, may expose these
lymphocytes in the GC environment to a high risk of transformation. Based on
normal Tfh biology and pathological data from AITL, we predict that signaling
pathways mediated by ICOS/ICOS-PI3K or SAP/SLAM family receptors may be crucial
for neoplastic expansion and spreading of AITL. Importantly, there are multiple
clinical trials targeting ICOS/ICOSL [71], PI3K isoforms [72], and SLAM family
receptors [73] to treat autoimmune diseases and cancer producing promising
results. Validation of these ideas in mouse models should galvanize new
approaches to treat AITL patients using these emerging drugs.
ACKNOWLEDGMENTS
This work is
supported by an operating grant from Cancer Research Society (W.-K. S.). M.W.
is a recipient of Graduate Scholarship from Canadian Institutes of Health
Research.
- Federico M., Rudiger T,
Bellei M., Nathwani BN, Luminari S, et al. (2013) Clinicopathologic
characteristics of angioimmunoblastic T-cell lymphoma: analysis of the
international peripheral T-cell lymphoma project. J Clin Oncol 31:
240-246.
- Xu B, Liu P (2014) No
survival improvement for patients with angioimmunoblastic T-cell lymphoma
over the past two decades: a population-based study of 1207 cases. PLoS
One 9: e92585.
- Crotty S (2014) T Follicular
Helper Cell Differentiation, Function, and Roles in Disease. Immunity 41:
529-542.
- Victora GD, Nussenzweig MC
(2012) Germinal centers. Annu Rev Immunol 30: 429-457.
- Pratama A, Vinuesa CG (2014)
Control of TFH cell numbers: why and how? Immunol Cell Biol 92: 40-48.
- Suh WK (2015) Life of T
Follicular Helper Cells. Mol Cells 38:195-201.
- de Leval L, Rickman DS,
Thielen C, Reynies A, Huang YL, et al. (2007) The gene expression profile
of nodal peripheral T-cell lymphoma demonstrates a molecular link between
angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH)
cells. Blood 109: 4952-4963.
- Piccaluga PP, Agostinelli C,
Califano A, Carbone A, Fantoni L, et al. (2007) Gene expression analysis
of angioimmunoblastic lymphoma indicates derivation from T follicular
helper cells and vascular endothelial growth factor deregulation. Cancer
Res 67: 10703-10710.
- Yu H, Shahsafaei A, Dorfman
DM (2009) Germinal-center T-helper-cell markers PD-1 and CXCL13 are both
expressed by neoplastic cells in angioimmunoblastic T-cell lymphoma. Am J
Clin Pathol 131: 33-41.
- Marafioti T, Paterson JC,
Ballabio E, Chott A, Natkunam Y, et al. (2010) The inducible T-cell
co-stimulator molecule is expressed on subsets of T cells and is a new
marker of lymphomas of T follicular helper cell-derivation. Haematologica
95: 432-439.
- Roncador G, Garcia
Verdes-Montenegro JF, Tedoldi S, Paterson JC, Klapper W, et al. (2007)
Expression of two markers of germinal center T cells (SAP and PD-1) in
angioimmunoblastic T-cell lymphoma. Haematologica 92: 1059-1066.
- de Leval L, Gisselbrecht C,
Gaulard P (2010) Advances in the understanding and management of
angioimmunoblastic T-cell lymphoma. Br J Haematol 148: 673-689.
- Sato F, Ishida T, Ito A, Mori
F, Masaki A, et al. (2013) Angioimmunoblastic T-cell lymphoma mice model.
Leuk Res 37: 21-27.
- Zhan HQ, Li XQ, Zhu XZ, Lu
HF, Zhou XY, et al. (2011) Expression of follicular helper T cell markers
in nodal peripheral T cell lymphomas: a tissue microarray analysis of 162
cases. J.Clin.Pathol 64: 319-324.
- Ellyard JI, Chia T,
Rodriguez-Pinilla SM, Martin JL, Hu X, et al. (2012) Heterozygosity for
Roquinsan leads to angioimmunoblastic T-cell lymphoma-like tumors in mice.
Blood 120: 812-821.
- Palomero T, Couronne L,
Khiabanian H, Kim MY, Ambesi-Impiombato A, et al. (2014) Recurrent
mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T
cell lymphomas. Nat Genet 46: 166-170.
- Sakata-Yanagimoto M, Enami T,
Yoshida K, Shiraishi Y, Ishii R, et al. (2014) Somatic RHOA mutation in
angioimmunoblastic T cell lymphoma. Nat Genet 46: 171-175.
- Yoo HY, Sung MK, Lee SH, Kim
S, Lee H, et al. (2014) A recurrent inactivating mutation in RHOA GTPase
in angioimmunoblastic T cell lymphoma. Nat Genet 46: 371-375.
- Lemonnier F, Couronne L,
Parrens M, Jais JP, Travert M, et al. (2012) Recurrent TET2 mutations in
peripheral T-cell lymphomas correlate with TFH-like features and adverse
clinical parameters. Blood 120: 1466-1469.
- Cairns RA, Iqbal J, Lemonnier
F, Kucuk C, de Leval L, et al. (2012) IDH2 mutations are frequent in
angioimmunoblastic T-cell lymphoma. Blood 119: 1901-1903.
- Cairns RA, Mak TW (2013)
Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and
clinical opportunities. Cancer Discov 3: 730-741.
- Palacios EH, Weiss A (2004)
Function of the Src-family kinases, Lck and Fyn, in T-cell development and
activation. Oncogene 23: 7990-8000.
- Yu D, Tan AH, Hu X,
Athanasopoulos V, Simpson N (2007) Roquin represses autoimmunity by
limiting inducible T-cell co-stimulator messenger RNA. Nature 450:
299-303.
- Heissmeyer V, Vogel KU (2013)
Molecular control of Tfh-cell differentiation by Roquin family proteins.
Immunol Rev 253: 273-289.
- Vinuesa CG, Cook MC,
Angelucci C, Athanasopoulos V, Rui L, et al. (2005) A RING-type ubiquitin
ligase family member required to repress follicular helper T cells and
autoimmunity. Nature 435: 452-458.
- Auguste T, Travert M, Tarte
K, Ame-Thomas P, Artchounin C, Martin-Garcia N, et al. (2013) ROQUIN/RC3H1
alterations are not found in angioimmunoblastic T-cell lymphoma. PLoS One
8: e64536.
- Basso K, Dalla-Favera R
(2015) Germinal centres and B cell lymphomagenesis. Nat Rev Immunol 15:
172-184.
- Basso K, Dalla-Favera R
(2012) Roles of BCL6 in normal and transformed germinal center B cells.
Immunol Rev 247: 172-183.
- Cattoretti G, Pasqualucci L,
Ballon G, Tam W, Nandula SV, et al. (2005) Deregulated BCL6 expression
recapitulates the pathogenesis of human diffuse large B cell lymphomas in
mice. Cancer Cell 7: 445-455.
- Baron BW, Anastasi J, Montag
A, Huo D, Baron RM, et al. (2004) The human BCL6 transgene promotes the
development of lymphomas in the mouse. Proc Natl Acad Sci USA 101:
14198-14203.
- Baron BW, Anastasi J, Hyjek
EM, Bies J, Reddy PL, et al. (2012) PIM1 gene cooperates with human BCL6
gene to promote the development of lymphomas. Proc Natl Acad Sci USA 109:
5735-5739.
- Baron BW, Anastasi J, Bies J,
Reddy PL, Joseph L, et al. (2014) GFI1B, EVI5, MYB--additional genes that
cooperate with the human BCL6 gene to promote the development of
lymphomas. Blood Cells Mol Dis 52: 68-75.
- Shaffer AL, Yu X, He Y,
Boldrick J, Chan EP, et al. (2000) BCL-6 represses genes that function in
lymphocyte differentiation, inflammation, and cell cycle control. Immunity
13: 199-212.
- Phan RT, Dalla-Favera R
(2004) The BCL6 proto-oncogene suppresses p53 expression in
germinal-centre B cells. Nature 432: 635-639.
- Okada T, Moriyama S, Kitano M
(2012) Differentiation of germinal center B cells and follicular helper T
cells as viewed by tracking Bcl6 expression dynamics. Immunol Rev 247:
120-132.
- Kitano M, Moriyama S, Ando Y,
Hikida M, Mori Y, et al. (2011) Bcl6 protein expression shapes
pre-germinal center B cell dynamics and follicular helper T cell
heterogeneity. Immunity 34: 961-972.
- Hutloff A, Dittrich AM, Beier
KC, Eljaschewitsch B, Kraft R, et al. (1999) ICOS is an inducible T-cell
co-stimulator structurally and functionally related to CD28. Nature 397:
263-266.
- Greenwald RJ, Freeman GJ,
Sharpe AH (2005) The B7 family revisited. Annu Rev Immunol 23: 515-548.
- Gigoux M, Shang J, Pak Y, Xu
M, Choe J, et al. (2009) Inducible costimulator promotes helper T-cell
differentiation through phosphoinositide 3-kinase. Proc Natl Acad Sci USA
106: 20371-20376.
- Rolf J, Bell SE, Kovesdi D,
Janas ML, Soond DR, et al. (2010) Phosphoinositide 3-kinase activity in T
cells regulates the magnitude of the germinal center reaction. J Immunol
185: 4042-4052.
- Xu H, Li X, Liu D, Li J,
Zhang X, et al. (2013) Follicular T-helper cell recruitment governed by
bystander B cells and ICOS-driven motility. Nature 496: 523-527.
- Grimbacher B, Hutloff A,
Schlesier M, Glocker E, Warnatz K, et al. (2003) Homozygous loss of ICOS
is associated with adult-onset common variable immunodeficiency. Nat
Immunol 4: 261-268.
- Warnatz K, Bossaller L,
Salzer U, Skrabl-Baumgartner A, Schwinger W, et al. (2006) Human ICOS
deficiency abrogates the germinal center reaction and provides a monogenic
model for common variable immunodeficiency. Blood 107: 3045-3052.
- Bossaller L, Burger J, Draeger
R, Grimbacher B, Knoth R, et al. (2006) ICOS deficiency is associated with
a severe reduction of CXCR5+CD4 germinal center Th cells. J Immunol 177:
4927-4932.
- Tafuri A, Shahinian A, Bladt
F, Yoshinaga SK, Jordana M, et al. (2001) ICOS is essential for effective
T-helper-cell responses. Nature 409: 105-109.
- McAdam AJ, Greenwald RJ,
Levin MA, Chernova T, Malenkovich N, et al. (2001) ICOS is critical for
CD40-mediated antibody class switching. Nature 409: 102-105.
- Dong C, Juedes AE, Temann UA,
Shresta S, Allison JP, et al. (2001) ICOS co-stimulatory receptor is
essential for T-cell activation and function. Nature 409: 97-101.
- Mak TW, Shahinian A,
Yoshinaga SK, Wakeham A, Boucher LM, et al. (2003) Costimulation through
the inducible costimulator ligand is essential for both T helper and B
cell functions in T cell-dependent B cell responses. Nat Immunol 4:
765-72.
- Linterman MA, Rigby RJ, Wong
R, Silva D, Withers D, et al. (2009) Roquin differentiates the specialized
functions of duplicated T cell costimulatory receptor genes CD28 and ICOS.
Immunity 30: 228-241.
- Okamoto T, Saito S, Yamanaka
H, Tomatsu T, Kamatani N, et al. (2003) Expression and function of the
co-stimulator H4/ICOS on activated T cells of patients with rheumatoid
arthritis. J Rheumatol 30: 1157-1163.
- Ruth JH, Rottman JB,
Kingsbury GA, Coyle AJ, Haines GK, et al. (2007) ICOS and B7 costimulatory
molecule expression identifies activated cellular subsets in rheumatoid
arthritis. Cytometry A 71: 317-326.
- Vogelzang A, McGuire HM, Yu
D, Sprent J, Mackay CR, et al. (2008) A fundamental role for
interleukin-21 in the generation of T follicular helper cells. Immunity
29: 127-137.
- Vogelzang A, McGuire HM, Liu
SM, Gloss B, Mercado K, et al. (2014) IL-21 contributes to fatal
inflammatory disease in the absence of Foxp3+ T regulatory cells. J
Immunol 192: 1404-1414.
- Gigoux M, Lovato A, Leconte
J, Leung J, Sonenberg N, et al. (2014) Inducible costimulator facilitates
T-dependent B cell activation by augmenting IL-4 translation. Mol Immunol
59: 46-54.
- Chen H, Fu T, Suh WK,
Tsavachidou D, Wen S, et al. (2014) CD4 T cells require ICOS-mediated PI3K
signaling to increase T-Bet expression in the setting of anti-CTLA-4
therapy. Cancer Immunol Res 2014. 2: 167-176.
- Guedan S, Chen X, Madar A,
Carpenito C, McGettigan SE, et al. (2014) ICOS-based chimeric antigen
receptors program bipolar TH17/TH1 cells. Blood 124: 1070-1080.
- Teichmann LL, Cullen JL,
Kashgarian M, Dong C, Craft J, et al. (2015) Local triggering of the ICOS
coreceptor by CD11c(+) myeloid cells drives organ inflammation in lupus.
Immunity 42: 552-565.
- Liu D, Xu H, Shih C, Wan Z,
Ma X, et al. (2014) T-B-cell entanglement and ICOSL-driven feed-forward
regulation of germinal centre reaction. Nature 10.
- Shulman Z, Gitlin AD,
Weinstein JS, Lainez B, Esplugues E,
et al. (2014) Dynamic signaling by T follicular helper cells during
germinal center B cell selection. Science 345: 1058-1062.
- Calpe S, Wang N, Romero X,
Berger SB, Lanyi A, et al. (2008) The SLAM and SAP gene families control
innate and adaptive immune responses. Adv Immunol 97: 177-250.
- Cannons JL, Tangye SG,
Schwartzberg PL (2011) SLAM family receptors and SAP adaptors in immunity.
Annu Rev Immunol 29: 665-705.
- Veillette A (2010)
SLAM-family receptors: immune regulators with or without SAP-family adaptors.
Cold Spring Harb Perspect Biol 2:
a002469.
- Morra M, Simarro-Grande M,
Martin M, Chen AS, Lanyi A, et al. (2001) Characterization of SH2D1A
missense mutations identified in X-linked lymphoproliferative disease
patients. J Biol Chem 276: 36809-36816.
- Davidson D, Shi X, Zhang S,
Wang H, Nemer M, et al. (2004) Genetic evidence linking SAP, the X-linked
lymphoproliferative gene product, to Src-related kinase FynT in T(H)2
cytokine regulation. Immunity 21: 707-717.
- Cannons JL, Yu LJ, Jankovic
D, Crotty S, Horai R, et al. (2006) SAP regulates T cell-mediated help for
humoral immunity by a mechanism distinct from cytokine regulation. J Exp
Med 203: 1551-1565.
- McCausland MM, Yusuf I, Tran
H, Ono N, Yanagi Y, et al. (2007) SAP regulation of follicular helper CD4 T
cell development and humoral immunity is independent of SLAM and Fyn
kinase. J Immunol 178: 817-828.
- Veillette A, Zhang S, Shi X,
Dong Z, Davidson D, et a. (2008) SAP expression in T cells, not in B
cells, is required for humoral immunity. Proc Natl Acad Sci USA 105:
1273-1278.
- Qi H, Cannons JL, Klauschen
F, Schwartzberg PL, Germain RN (2008) SAP-controlled T-B cell interactions
underlie germinal centre formation. Nature 455: 764-769.
- Cannons JL, Qi H, Lu KT,
Dutta M, Gomez-Rodriguez J, et al. (2010) Optimal germinal center
responses require a multistage T cell:B cell adhesion process involving
integrins, SLAM-associated protein, and CD84. Immunity 32: 253-265.
- Zhong MC, Veillette A (2013)
Critical role of SAP in progression and reactivation but not maintenance
of T cell-dependent humoral immunity. Mol Cell Biol 33: 1223-1232.
- Merrill JT (2013)
Co-stimulatory molecules as targets for treatment of lupus. Clin Immunol
148: 369-375.
- Fruman DA, Rommel C (2014)
PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug
Discov 13: 140-156.
- Veillette A, Guo H (2013)
CS1, a SLAM family receptor involved in immune regulation, is a
therapeutic target in multiple myeloma. Crit Rev Oncol Hematol 88:
168-177.
QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Ophthalmology Clinics and Research (ISSN:2638-115X)
- Journal of Cell Signaling & Damage-Associated Molecular Patterns
- International Journal of Anaesthesia and Research (ISSN:2641-399X)
- Dermatology Clinics and Research (ISSN:2380-5609)
- Journal of Clinical Trials and Research (ISSN:2637-7373)
- Journal of Cardiology and Diagnostics Research (ISSN:2639-4634)
- Journal of Alcoholism Clinical Research



