642
Views & Citations10
Likes & Shares
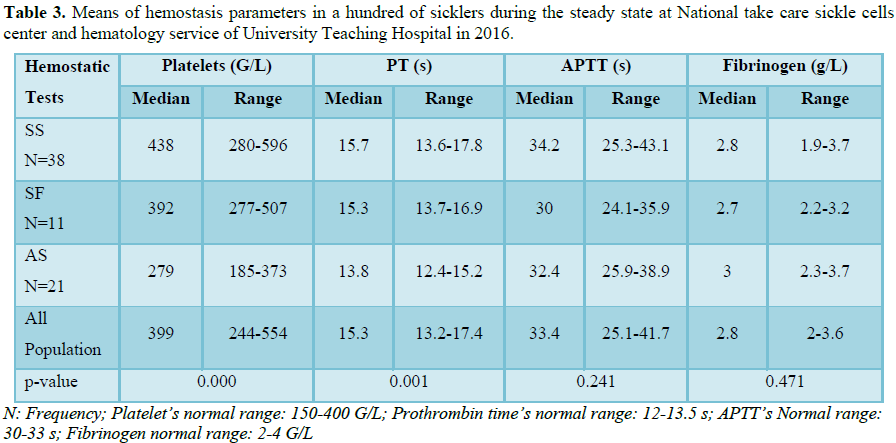
Sickle-cell anemia is characterized by a hypercoagulable state with a perturbed coagulation profile during vaso-occlusive crises, as well as during inter-critical periods. Some studies have been done in several countries on the coagulation profile during inter-critical periods in patients suffering from this hemoglobinopathy, but there are no reports on Cameroonian sickle-cell patients during the inter-critical period. In this prospective study, we included 68 blood samples homozygous SS sickle-cell patients, 11 samples from heterozygous patients (SF patients and 21 AS patients). Standard techniques were used for the measurements of blood counts, prothrombin time, activated partial thromboplastin time and fibrinogen levels in all samples. The study was carried out from February to August 2016. The mean platelet count in homozygous SS patients was 438 ± 158 G/L and was significantly higher than that of AS samples (279 ± 94 G/L, p=0.000) and of SF samples (392 ± 115 G/L, p=0.037). The mean prothrombin time was 15.7 ± 2.1 seconds (s) for SS samples and was significantly different from that of AS (13.8 ± 1.4 s, p=0.000) samples but not significantly different from that of SF samples (15.3 ± 1.6 s, p=0.525). Activated partial thromboplastin time and mean fibrinogen levels in SS samples (34.2 ± 8.9 s and 2.8 ± 0.9 g/L, respectively) were not statistically different from those of AS samples (32.4 ± 6.5 s, p=0.373 and 3 ± 0.7 g/L, p=0.240, respectively) and of SF samples (30 ± 5.9 s, p=0.436 and 2.7 ± 0.5 g/L, p=0.359, respectively).
The coagulation profile of Cameroonian sickle cell sufferers in the inter-critical period shows more abnormal values in SS and SF patients than in AS patients. However, the prothrombin time, activated partial thromboplastin time and fibrinogen levels remain within normal limits.
Keywords: Coagulation, Sickle-cell anemia, Platelets, Prothrombin time
Abbreviations: APTT: Activated Partial Thromboplastin Time; g/L: grams per liter; G/L: 1003 per mm3, s: Seconds; PT: Prothrombin Time
INTRODUCTION
Sickle Cell Anemia is a hereditary hemoglobinopathy transmitted in autosomal recessive fashion. It results from a mutation of the beta-globin chain gene on chromosome 11, a mutation in which glutamic acid is replaced by valine in position 6 [1]. It is characterized by a hypercoagulable state in which several perturbations during and in-between vaso-occlusive crises cause an increase in the activation of the coagulation system, platelets, prothrombin time and thus occurrence of thrombosis [2-5]. Decreased natural anticoagulant proteins C and S [6], increased prothrombin time and hyperfibrinogenemia have also been reported in the absence of vaso-occlusive crises.
MATERIALS AND METHODS
The study was carried out at the National Center for the Management of Sickle-Cell Anemia, the sickle-cell anemia unit of the Chantal Biya Foundation and the Hematology Laboratory of the University Teaching Hospital in Yaoundé, over a period of seven months. Stable patients were SS and SF patients who were without pain necessitating pain-killers for a minimum period of four weeks [1], between the last day of the previous crisis and the first day of the next vaso-occlusive crisis. Carriers of sickle-cell trait were also included in the study.
With the aid of a pre-conceived data-collection form, demographical (age, sex), hematological (platelet counts, hemoglobin electrophoresis) and hemostatic parameters (PT, APTT, fibrinogen levels) of each subject were recorded. For each subject, two venous blood samples were collected in EDTA and citrated tubes. Complete blood counts were done in EDTA tubes with MINDRAY BC 5300 analyzer (Medical Electronics Technology Research Institute Co., Ltd., Shenzen, China) within 3 h following blood collection. Samples collected in the citrated tubes were used for coagulation tests (PT, APTT, fibrinogen levels). The samples were centrifuged at 1200 rpm for 15 min to obtain platelet-poor plasma, which was introduced into a Stago START 4 coagulation analyzer, version 2002, from France for haemostatic tests. Thromboplastin (Neoplastin®) used for measurement of prothrombin time and fibrinogen (Fibrinogène®) were obtained from Cypress Diagnostics while C.K. Prest® was used for measurement of APTT (supplied by Stago, France). All coagulation tests were done in duplicates for each plasma sample between two to four hours following the collection. Results were expressed in terms of means (in seconds, s) for PT and APTT. Fibrinogen levels were measured in grams per liter (g/L).
Ethical clearance and authorizations for research were obtained from the participating centres and the national review board.
Quantitative variables were analyzed as continuous variables and means were used to evaluate central tendencies and distribution. Descriptive analysis of demographic, hematological and hemostatic parameters between the subgroups was also carried out. Data were entered in CSPRO version 6.1 software and statistical analysis was done with SPSS 16.0. Means and standard deviations were compared using ANOVA tests. The Student T test enabled us to compare the groups two-by-two. Statistical significance was set at p<0.05.
RESULTS AND DISCUSSION
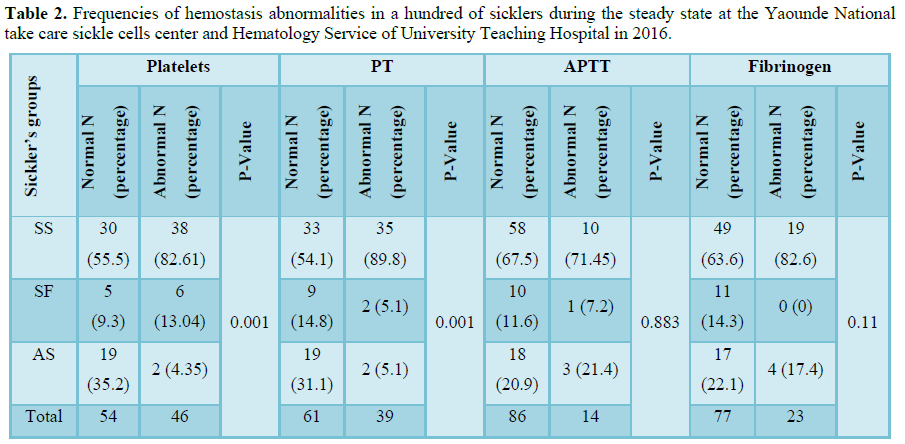
One hundred blood samples were collected from sickle-cell subjects aged 2 to 56 years predominantly the Sudanese ethnic group. The homozygous sicklers represented 68% of the participants (Table 1). A total of 82.6% homozygous had an abnormal number of platelets and 89.6% had an abnormal prothrombin time. Thrombocytosis was found only in SS (Table 2). The means of platelets and prothrombin time of SS sicklers were 438 G/L and 15.7 s, respectively in homogenous sicklers compared to 279 G/L, p=0.000 and 13.8 s; p=0.000, respectively in heterogenous sicklers AS and 392 G/L; p=0.037 and 15.3 s; p=0.525 in SF. Our study showed that hemostatic abnormalities in Cameroonian sickler are predominantly an increase of platelet count in eight patients over ten, homozygous are more affected than heterogenous and a prolonged prothrombin time is present in half the patients (Table 3).
The raised platelet count in sicklers has previously been reported with a high tendency of homozygotes to have thrombocytosis [8,9]. This thrombocytosis is probably a consequence of the loss of the function of splenic sequestration which results from autosplenectomy in these patients. The spleen is the main site of destruction of platelets so in the event of splenectomy reactive thrombocytosis occurs. According to Girot et al. [8], this thrombocytosis could be a manifestation of hyposplenism.
LIMITATIONS
We recognize some limitations in this study. Measurement of coagulation factors and transaminases was not done in our study but this does not void the validity of our results. However our sample size should be enlarged to confirm these results.
CONCLUSION
Based on our study, Cameroonian sicklers during steady state have a prolonged prothrombin time and increase in platelets’ count. Future larger prospective studies to determine whether abnormal coagulation findings in sicklers are associated with a decrease in coagulation factors and transaminases are necessary in our context.
Just like the complete blood count, prothrombin time measurement should become a routine test in the evaluation of sickle cell anemia patients in order to evaluate their liver function. The early detection of liver dysfunction amongst these patients will permit that they are managed adequately.
1. Rahimi Z, Parsian A (2011) Sickle cell disease and venous thromboembolism. Mediterr J Hematol Infect Dis 3: e2011024.
2. Ataga KI, Cappellini MD, Rachmilewitz EA (2007) Beta-thalassemia and sickle cell anemia as paradigms of hypercoagulability. Br J Hematol 139: 3-13.
3. Ataga KI, Orringer EP (2003) Hypercoagulability in sickle cell disease: A curious paradox. Am J Med 115: 721-728.
4. Rahimi Z, Vaisi-Raygani A, Nagel RL, Muniz A (2008) Thrombophilic mutations among Southern Iranian patients with sickle cell disease: High prevalence of factor V Leiden. J Thromb Thrombolysis 25: 288-292.
5. Francis RB (1991) Large-vessel occlusion in sickle cell disease: Pathogenesis, clinical consequences and therapeutic implications. Med Hypotheses 35: 88-95.
6. Westerman MP, Green D, Gilman-Sachs A, Beaman K, Freels S, et al. (2002) Coagulation changes in individuals with sickle cell trait. Am J Hematol 69: 89-94.
7. Behrman RE, Kliegman RM, Jenson HB (2003) Nelson Textbook of Pediatrics. (17th Edn), United States of America: Judith Fletcher, pp: 1623-1634.
8. Girot R (2017) Hématologie des syndromes drépanocytaires In: La maladie drépanocytaire. (Sandoz Ed.), Paris, France: In: Begue, pp: 64-73.
9. Izuwa G, Akpotuzor JO, Okpokam DC, Akpan PA, Ernest NA, et al. (2016) Haemorrheologic and fibrinolytic activities of HbSS, HbAS and HbAA subjects in Abuja, Nigeria. J Med Sci 16: 32-37.
10. Raffini LJ, Niebanck AE, Hrusovsky J, Stevens A, Blackwood-Chirchir A, et al. (2006) Prolongation of the prothrombin time and activated partial thromboplastin time in children with sickle cell disease. Pediatr Blood Cancer 47: 589-593.
11. Chinawa JM, Emodi IJ, Ikefuna AN, Ocheni S (2013) Coagulation profile of children with sickle cell anemia in steady state and crisis attending the university of Nigeria teaching hospital, Ituku-Ozalla, Enugu. Niger J Clin Pract 16: 159-163.
12. Embury SH (1986) The clinical pathophysiology of sickle cell disease. Annu Rev Med 37: 361-376.
13. Winichagoon P, Fucharoen S, Wasi P (1981) Increased circulating platelet aggregates in thalassemia. Southeast Asian J Trop Med Public Health 12: 556-560.
14. Wright JG, Malia R, Cooper P, Thomas P, Preston FE, et al. (1997) Protein C and protein S in homozygous sickle cell disease: Does hepatic dysfunction contribute to low levels? Br J Haematol 98: 627-631.
-
 Table 1
Table 1 -
Table 2
-
Table 3

QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Chemotherapy Research Journal (ISSN:2642-0236)
- Journal of Psychiatry and Psychology Research (ISSN:2640-6136)
- Journal of Otolaryngology and Neurotology Research(ISSN:2641-6956)
- International Journal of Diabetes (ISSN: 2644-3031)
- Journal of Allergy Research (ISSN:2642-326X)
- International Journal of Internal Medicine and Geriatrics (ISSN: 2689-7687)
- Journal of Infectious Diseases and Research (ISSN: 2688-6537)


