5186
Views & Citations4186
Likes & Shares
E-PodoFavalin-15999
(Atremorine®) is a novel biopharmaceutical compound, obtained by means of
non-denaturing biotechnological procedures from structural components of Vicia faba L., for the prevention and
treatment of Parkinsonian disorders. Preclinical studies revealed that
Atremorine is a powerful neuroprotectant with specific activity on dopaminergic
neurons, reversing neurodegeneration and improving motor function in animal
models of Parkinson’s disease (PD).
This is
the first clinical study in Parkinsonian patients (N=119) addressing
Atremorine-induced dopamine response. One hour after a single oral dose of
Atremorine (5g), plasma DA levels increased from 762.28 ± 296.94 to 4556.61 ±
678.95 pg/mL in the whole group (p<0.001). In patients never treated before
with antiparkinsonian drugs, DA levels increased from 11.22 ± 0.29 to 2041.24 ±
249.12 pg/mL (p<0.001), with a response rate of 100%; and in patients chronically treated with
anti-PD drugs, DA levels raised from 2139.23 ± 804.72 to 9168.11 ± 1657.27
pg/mL (p<0.001) with a response rate of 98%. No significant differences in
the magnitude of the response were observed between females and males.
The
Atremorine-induced dopamine response was different in carriers of APOE and CYP
variants. APOE-2 carriers showed a stronger response than APOE-3>APOE-4
carriers. Although a significant 200-500-fold increase in DA levels was common
in over 80% of patients, CYP2D6-, CYP2C19-, CYP2C2- and CYP3A4/5-EMs and IMs showed
a better response than PMs and UMs.
Atremorine
is a powerful pro-dopaminergic neuroprotectant with potential preventive and
therapeutic effects in neurodegenerative disorders that compromise the
dopaminergic system.
Keywords: Atremorine, Dopamine, APOE, CYPs, Parkinson’s
disease, Pharmacogenetics
INTRODUCTION
Parkinson’s
disease (PD) is the second most important neurodegenerative disorder in the
elderly population, after Alzheimer’s disease. With a prevalence ranging from
35.8 per 100,000 to 12,500 per 100,000 and annual incidence estimates ranging
from 1.5 per 100,000 to 346 per 100,000 in different countries [1-3], PD is
becoming a major age-related problem of health [4,5]. Meta-analysis of the
worldwide data indicate a rising prevalence of PD with age (41 per 100,000 in
40-49 years; 107 in 50-59 years; 173 in 55-64 years; 428 in 60-69 years; 425 in
65-74 years; 1087 in 70-79 years; and 1903 per 100,000 in older than age 80),
also reflecting a characteristic distribution by geographic location (a
prevalence of 1,601 per 100,000 in patients from North America, Europe and
Australia, and a prevalence of 646 per 100,000 in Asian patients) [6]. PD is
more prevalent in males (1729 per 100,000, >65 yrs) than in females (1644
per 100,000), with a peak prevalence in the age group of ≥90 years (4633 cases
per 100,000), and a mean prevalence of 1680 per 100,000 in people older than 65
years of age [7]. Prevalence and incidence Male/Female ratios increase by 0.05
and 0.14, respectively, per 10 years of age. Incidence is similar in men and
women under 50 years (M/F ratio <1.2), and over 1.6 times higher in men than
women above 80 years [8].
Furthermore, PD coexists with dementia in over 25% of the cases and with
depression in over 30% of the cases in some countries [7].
Associated with different potentially pathogenic risk factors (toxins,
drugs, pesticides, brain microtrauma, focal cerebrovascular damage, genomic
defects), PD neuropathology is characterized by a selective loss of
dopaminergic neurons in the substantia nigra pars compacta, with widespread involvement
of other CNS structures and peripheral tissues. PD-related neurodegeneration is
likely to occur several decades before the onset of the motor symptoms (rigidity,
bradykinesia, resting tremor) [9].
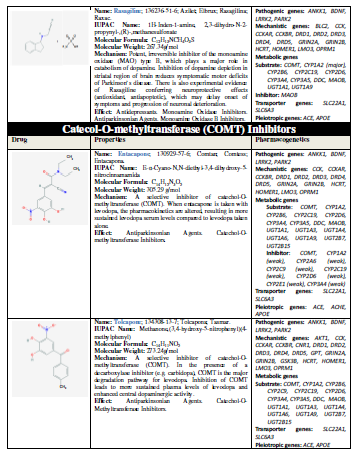
The introduction of L-DOPA in the 1960s represented a breakthrough in
the treatment of PD, and it continues to be the most effective symptomatic
therapy in Parkinsonian disorders [10]. In addition to dopamine precursors
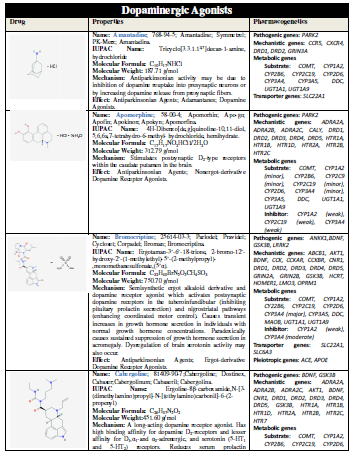
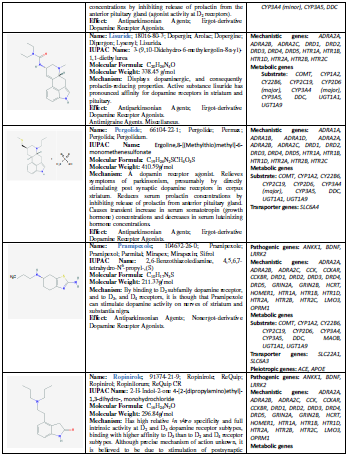
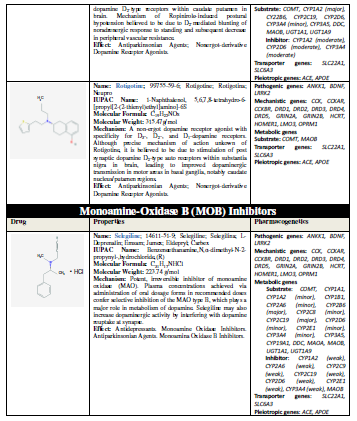
(L-DOPA), other symptomatic treatments for PD include dopamine agonists
(amantadine, apomorphine, bromocriptine, cabergoline, lisuride, pergolide,
pramipexole, ropinirole, rotigotine), monoamine oxidase (MAO) inhibitors
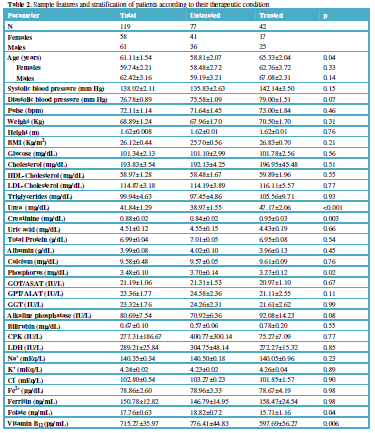
(selegiline, rasagiline), and catechol-O-methyltransferase (COMT) inhibitors
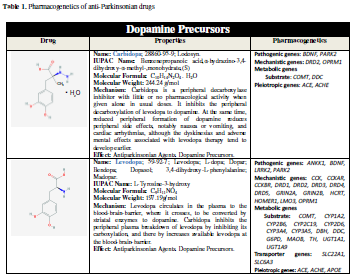
(entacapone, tolcapone) [11] (Table 1).
The initial complication of long-term L-DOPA therapy is the “wearing-off”
phenomenon [12,13], together with motor
fluctuations and dyskinesia which develop during the use of both L-DOPA and
dopamine agonists [10,14]. Diverse dopaminergic and nondopaminergic
pharmacological approaches have been developed to manage such complications,
including novel L-DOPA formulations, COMT inhibitors (opicapone), dopamine
agonists, adenosine A2A antagonists (istradefylline, preladenant, tozadenant),
glutamatergic N-methyl-d-aspartate (NMDA) antagonists, serotonergic agents
(eltoprazine), and glutamate mGluR5 modulators (mavoglurant), with
controversial results [15,16]. Polypharmacy with antidepressants,
antipsychotics, urological drugs, analgesics, antihistaminics and
cholinesterase inhibitors also contributes to severe complications associated
with the anticholinergic burden in PD [17].
Furthermore, gastrointestinal complications (constipation, sialorrhea,
dysphagia, difficulty in mastication, choking/aspiration) [18], cardiovascular
problems [19], neuroendocrine changes and psychiatric disorders are frequent in
PD patients chronically treated with conventional antiparkinsonian drugs
[11,18].
We introduce here, for the first time, E-PodoFavalin-15999
(Atremorine®), a novel biopharmaceutical compound, obtained by means of
non-denaturing biotechnological procedures from structural components of Vicia faba L., for the prevention and
treatment of PD [20]. Preclinical studies (in vitro) revealed that Atremorine
is a powerful neuroprotectant in (i) cell cultures of human neuroblastoma
SH-SY5Y cells; (ii) hippocampal slices in conditions of oxygen and glucose
deprivation; and (iii) striatal slices under conditions of neurotoxicity
induced by 6-OHDA. In vivo studies showed that Atremorine (i) protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
(MPTP)-induced dopaminergic
neurodegeneration; (ii) inhibits MPTP-induced microglia activation and
neurotoxicity in substantia nigra; and (iii) improves motor function in mice
with MPTP-induced neurodegeneration [20,21]. Clinical studies in untreated patients who receive Atremorine for the
first time (never treated before with antiparkinsonian drugs) revealed that
Atremorine enhances dopaminergic neurotransmission and increases by
200-500-fold plasma dopamine levels. In patients chronically treated with L-DOPA
or other antiparkinsonian drugs, Atremorine induces a dopamine response of
similar magnitude to that observed in previously untreated patients. Atremorine
is also a powerful regulator of noradrenaline and pituitary hormones such as
prolactin and growth hormone, which are under supra-hypothalamic control of
dopaminergic neurotransmission. In addition, this dopaminergic response is
associated with the pharmacogenetic profile of the patients [20].
MATERIAL
AND METHODS
Patients and
Treatment
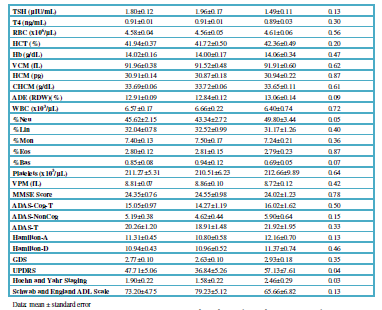
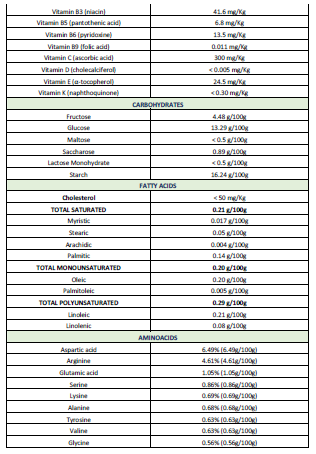
Patients (N=119; age: 61.11 ± 1.54 yrs) of both sexes (58 Females, age:
59.74 ± 2.21; 61 Males, age: 62.42 ± 3.16 yrs) with Parkinsonian disorders
(Idiopathic PD, 49; Hemiparkinsonism, 4; Vascular PD, 24; Post-traumatic PD,
10; Toxic PD, 10; Parkinson-Dementia Complex, 13; Congenital Extrapyramidal
syndrome, 5; Cadasil-associated PD, 1; Familial PD, 3) were recruited for this
study. The selected patients were divided into two groups: (i) Untreated
patients (U; N=77, age: 58.81±2.07 yrs), who had never received any antiparkinsonian
drug before; and (ii) patients chronically treated (T) with L-DOPA and other
antiparkinsonian drugs (N=42, age: 65.33±2.04 yrs) (Table 2). All patients underwent, under informed consent, the following protocol: (i) Clinical
(neurologic, psychiatric) examination, (ii) blood and urine analyses (Table 2), (iii) neuropsychological
assessment (MMSE, ADAS, Hamilton-A/D, GDS, UPDRS, Hoehn and Yahr Staging,
Schwab and England ADL Scale) (Table 2),
(iv) cardiovascular evaluation (EKG), (v) structural neuroimaging (brain MRI),
(vi) functional neuroimaging (brain mapping, brain optical topography), (vii)
genetic assessment (APOE), and (viii) pharmacogenetic profiling (CYP2D6,
CYP2C19, CYP2C9, CYP3A4/5).
All patients received a single oral dose of
5g E-PodoFavalin-15999 (Atremorine®) (Table
3) in the morning to avoid circadian variations in biochemical and hormonal
parameters, and blood samples were obtained prior to Atremorine intake and 60
minutes later.
Analytical
methods
Venous blood samples were taken after
overnight fasting with patients in supine position. Blood was collected in BD
Vacutainer serum separation tubes while blood for analysis of plasma dopamine
was collected in EDTA containing tubes. Specimens for dopamine analysis were immediately
placed on ice and centrifuged at 3000 rpm, at 4°C, for 10 minutes, soon after
venous extraction [22]. Serum tubes were allowed to clot at room temperature
during 30 minutes before processing and were centrifuged within 60 minutes of
sampling under the same conditions as the EDTA tubes. After refrigerated
centrifugation serum and plasma were removed from blood cells [23] and placed
in an appropriate sample container. Plasma aliquots for fractionated dopamine
determination were stored at -20 °C for no more than one week and purified with
albumin until their analysis by High
Performance Liquid Chromatography (HPLC) with electrochemical detection [24,25]. The HPLC system consisted of pump (515
Waters, USA), autosampler (717 Waters, USA), chromatographic column (Resolve
C18 Waters, USA), electrochemical detector (2465 Waters, USA) and Empower2
chromatography data software (Waters, USA).
Genotype
analysis
DNA was extracted from peripheral blood using Qiagen extraction columns (Qiagen, Hilden, Germany). A total of 13 single nucleotide polymorphisms (SNPs) and 1 copy number variation polymorphism (CNV) from 6 different genes (Table 4) were genotyped. APOE ε2, ε3, and ε4 alleles were defined by SNPs rs429358 (3932T>C Cys112Arg) and rs7412 (4070C>T, Arg158Cys). CYP2D6 alleles were identified as *1 (wild type), *1xN (gene duplication), *3 (rs35742686, 775delA, Arg259Glyfs), *4 (rs3892097, 506-1G>A), *5 (gene deletion), *6 (rs5030655, 454delT, Trp152Glyfs) and *41 (rs28371725, 985+39G>A). CYP2C9 alleles were *1 (wild type), *2 (rs1799853, 430C>T, Arg144Cys) and *3 (rs1057910, 1075A>C, Ile359Leu). CYP2C19 alleles were *1 (wild type), *2 (rs4244285, 681G>A, Pro227Pro) and *17 (rs12248560, -806C>T). CYP3A4 alleles were *1 (wild type), *1G (rs2242480, 1026+12G>A) and *22 (rs35599367, 522-191C>T). CYP3A5 alleles were *1 (wild type), *3 (rs776746, 219-237G>A). RT-PCR amplification (Real-Time Polymerase Chain Reaction) was performed using TaqMan assays for SNPs using StepOne Plus Real Time PCR System (Life Technologies, Waltham, Massachusetts, USA) and/or TaqMan®OpenArray® DNA microchips for QuantStudioTM 12K Flex Real-Time PCR System. OpenArray® genotyping analysis was performed using the Genotyper software (Thermo Fisher Scientific, Waltham, Massachusetts, USA).
Statistical analysis
Data were analyzed by using IBM SPSS Statistics 20 and SigmaPlot 10.0 Software. Comparisons between groups were studied by t-Test, Mann-Whitney Rank Sum Test, Chi Square without Yates correction and Fisher exact, and Pearson Correlation Analysis (Nonlinear Regression, Durbin-Watson Statistic, Normality Test, Constant Variance Test, 95% Confidence). All values are expressed as mean ± SE, and the degree of significance is considered when p<0.05.
RESULTS
Basal dopamine
levels
Atremorine was well tolerated by 100% of patients, and no side effects
were reported in either U or T patients. Clinical improvement lasted for 3 to
12 hrs in U patients.
Basal DA levels in the whole group were 762.28 ± 296.94 pg/mL
(range:8-30318 pg/mL), and were lower in females (232.05 ± 107.33 pg/mL) than
in males (1266.44 ± 564.98 pg/mL) (p=0.03). Drastic differences were seen in
basal DA levels between untreated patients (U) (11.22 ± 0.29 pg/mL) and
patients chronically treated with antiparkinsonian drugs (T)(2139.23±804.72
pg/mL) (p<0.001). Basal DA levels in U patients were below 20 pg/mL in
practically 100% of the cases with a clear homogeneity; however, in T patients
DA levels were extremely variable, ranging from >20 to 30318 pg/mL).
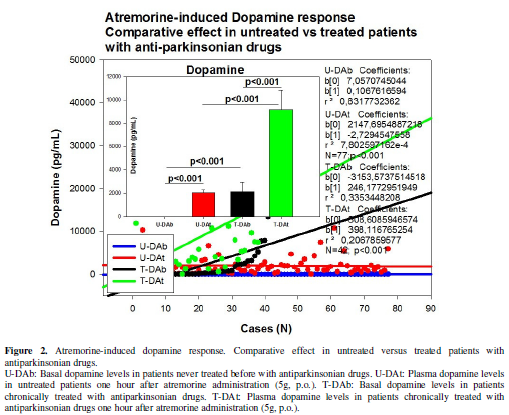
Atremorine-induced
dopamine response
A single oral dose of Atremorine (5g) induced an increase in DA levels
up to 4556.61 ± 678.95 pg/mL (p<0.001) (Figure
1). In U patients DA levels increased from 11.22 ± 0.29 to 2041.24 ± 249.12
pg/mL (p<0.001), with a response rate of 100%, and in T patients DA levels
rose from 2139.23 ± 804.72 to 9168.11±1657.27 pg/mL (p<0.001) after one hour
(Figure 2), with a response rate of
98% (Figure 2). No significant
differences in the magnitude of the response were observed between females and
males.
Pharmacogenetics of Atremorine-induced
Dopamine response
Plasma DA response to Atremorine was in part associated with the APOE
genotype of patients as well as with their pharmacogenetic profile. Basal DA
levels were substantially different among APOE-2 (294.89 ± 155.92 pg/mL),
APOE-3 (752.20 ± 314.20 pg/mL) and APOE-4 allele carriers (2121.63 ± 1212..97
pg/mL), with significant differences between APOE-2 and APOE-4 carriers
(p<0.05); however, APOE allele-related DA surge was similar in APOE-2
(7765.36 ± 2040.83 pg/mL), APOE-3 (4469.67 ± 717.18 pg/mL) and APOE-4 carriers
(5434.77 ± 1830.97 pg/mL), although the magnitude of the response with regard
to basal levels was the strongest in APOE-2 carriers and weaker in APOE-4 carriers.
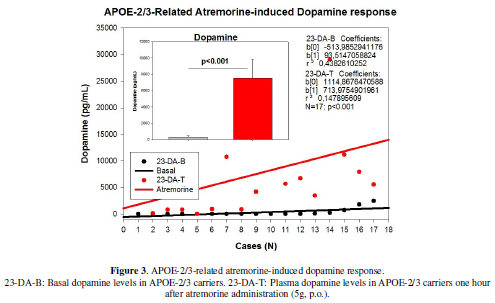
The distribution and frequency of APOE genotypes were as follows:
APOE-2/2 0%, APOE-2/3 14.53%, APOE-2/4 1.71%, APOE-3/3 58.12%, APOE-3/4 25.64%,
and APOE-4/4 0% (Table 5). DA levels
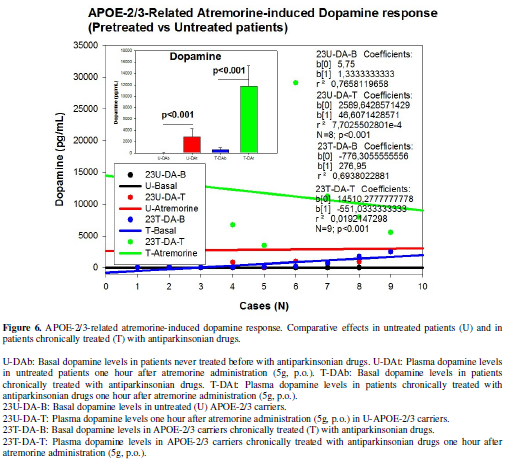
increased from 327.64 ± 173.00 to 7540.64 ± 2273.79 pg/mL in APOE-2/3 carriers
(p<0.001) (Figure 3); from 16.50 ±
4.50 to 9675.50 ± 2236.50 pg/mL in 2 cases harboring the APOE-2/4 genotype;
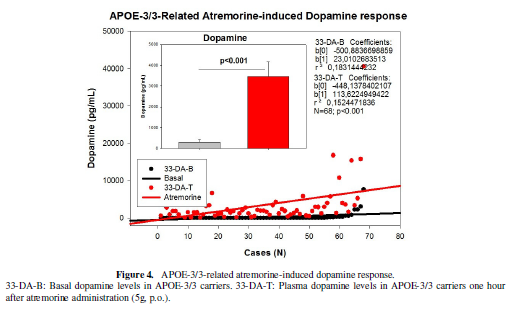
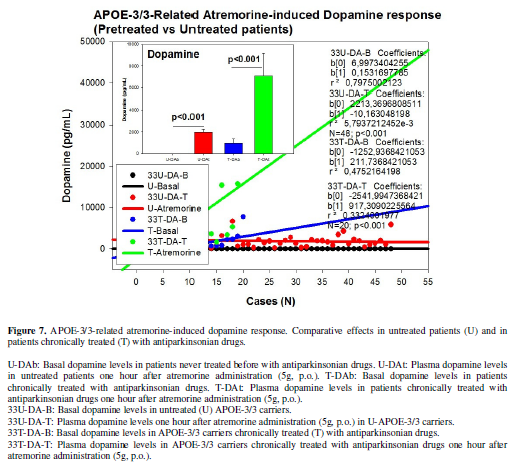
from 292.97 ± 128.93 to 3471.83 ± 697.81 pg/mL in APOE-3/3 carriers (p<0.001)
(Figure 4); and from 2290.40 ± 1305.93
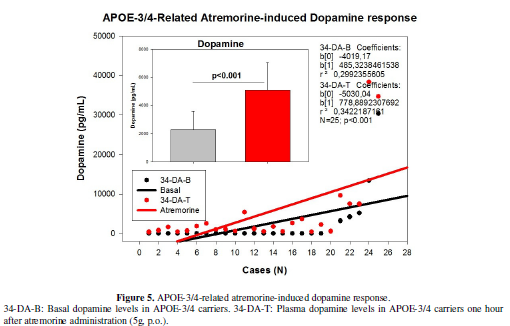
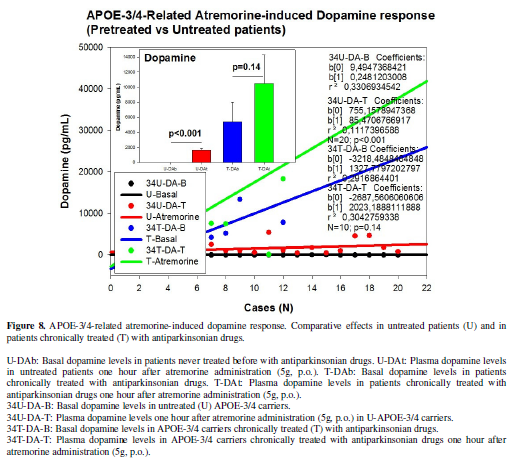
to 5095.52 ± 1959.83 pg/mL (p<0.001) in APOE-3/4 carriers (Figure 5). Significant differences were
found between U and T patients according to their APOE genotype (Figure 6-8). DA levels in U APOE-2/3
patients increased from 11.75±1.31 to 2799.37±303.52 pg/mL (p<0.001); and
from 608.44 ± 303.52% to 11755.00 ± 3628.85 pg/mL (p<0.001) in T patients (Figure 6). In U APOE-3/3 carriers DA
levels increased from 10.75±0.34 to 1964.37±269.80 pg/mL (p<0.001), and in T
APOE-3/3 carriers DA levels augmented from 970.30 ± 406.32 to 7089.75±2104.76pg/mL
(p<0.001) (Figure 7). In U
APOE-3/4 carriers DA levels changed from 12.10 ± 0.57 to 1652.60±338.24 pg/mL
(p<0.001), whereas T APOE-3/4 carriers responded to Atremorine with an
increase in DA levels from 5412.08 ± 2558.37 to 10463.16 ± 3817.54 (p=0.14) (Figure 8).
Important differences were also observed in DA response to Atremorine
in patients with different metabolizing enzyme capacity associated with CYP2D6,
CYP2C19, CYP2C9 and CYP3A4/5 genotypes, according to their condition of extensive
(EM), intermediate (IM), poor (PM), rapid (RM) or ultra-rapid metabolizers (UM)
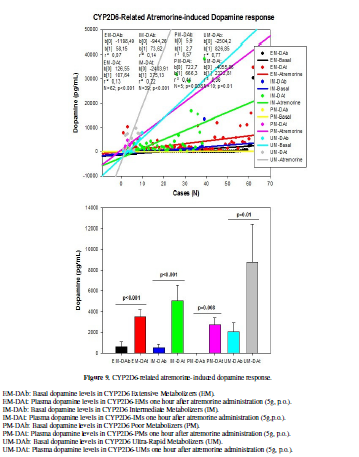
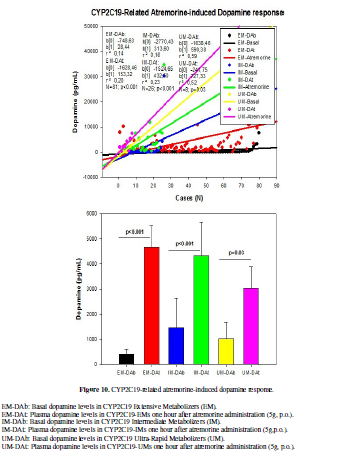
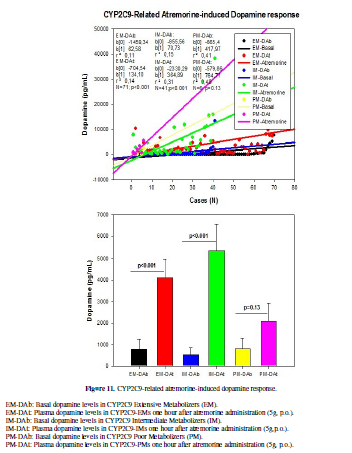
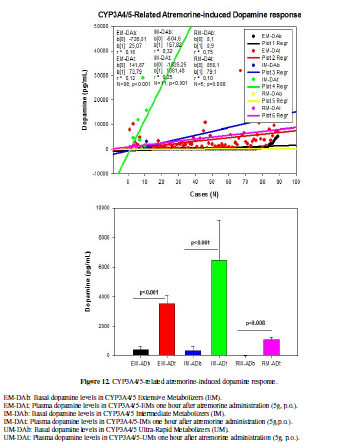
(Figure 9-12).
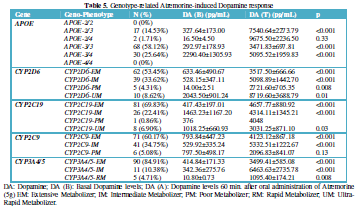
CYP2D6 geno-phenotypes were as follows: EMs 53.45%, IMs 33.62%, PMs
4.31%and UMs 8.62% (Table 5). DA
levels increased from 633.46 ± 490.67 to 3517.50 ± 666.66 pg/ml (p<0.001) in
CYP2D6-EMs, from 528.15 ± 347.11 to 5098.87 ± 1441.70 pg/mL (p<0.001) in
CYP2D6-IMs, from 14.00 ± 2.51 to 2721.60 ± 705.35 pg/mL (p=0.008) in
CYP2D6-PMs, and from 2043.50 ± 901.24 to 8719.60 ± 3688.79 pg/mL (p=0.01) in
CYP2D6-UMs (Table 5, Figure 9).
CYP2C19 geno-phenotypes were 69.83%, 22.41%, 0.86% and 6.90% for EMs,
IMs, PMs and UMs, respectively (Table
5). CYP2C19-EMs showed an increase in DA levels from 417.43 ± 197.01 to
4657.77 ± 880.92 pg/ml (p<0.001), whereas in CYP2C19-IMs and UMs, DA levels
increased from 1463.23 ± 1167.20 to 4314.11 ± 1345.21 pg/mL (p<0.001), and
from 1018.25 ± 660.93 to 3031.25 ± 871.10 pg/mL (p=0.03), respectively (Figure 10).
The frequency of CYP2C9-EMs, IMs and PMs were 60.17%, 34.75% and 5.06%,
respectively. In CYP2C9-EMs, DA levels raised from 793.84 ± 447.23 to 4123.12 ±
867.18 pg/mL (p<0.001). CYP2C9-IMs exhibited an increase in DA levels from
529.92 ± 335.24 to 5332.51 ± 1222.67 pg/mL (p<0.001); however, this
response, though quantitatively important (from 797.50±498.17 to 2096.83±841.07
pg/mL), was not significant (p=0.13) in CYP2C9-PMs (Table 5, Figure 11).
DA levels
in CYP3A4/5-EMs (84.91%) increased from 414.84 ± 171.33 to 3499.41 ± 585.08
pg/mL (p<0.001). In CYP3A4/5-IMs (10.38%) DA levels increased from 342.36 ±
275.76 to 6463.63 ± 2735.78 pg/mL (p<0.001); and in CYP3A4/5-RMs DA levels
changed from 10.80 ± 0.73 to 1095.40 ± 174.21 pg/mL (p=0.008) one hour after
Atremorine intake (Table 5, Figure 12).
DISCUSSION
This first clinical study with Atremorine in patients with Parkinsonian disorders clearly demonstrates the powerful effect of this novel bioproduct on plasma dopamine (Figure 1) in both untreated patients and patients chronically treated with conventional antiparkinsonian drugs (Figure 2). This pro-dopaminergic effect can be attributed to the rich content of natural L-DOPA (average concentration 20 mg/g) in the composition of Atremorine (Table 2). However, the neuroprotective effect of this nutraceutical product on dopaminergic neurons, as demonstrated in in vitro studies [20] and in animal models of PD [21], cannot be only attributed to L-DOPA, but to other intrinsic constituents (selective neurotrophic factors) of the compound [20].This study also makes clear that 100% of untreated PD patients exhibit a dramatic hypodopaminemia, with plasma levels of DA below 20 pg/mL (Table 5) and that PD patients under long-term treatment with L-DOPA and/or conventional antiparkinsonian drugs experience a hyperdopaminemic status which might be responsible for (i) the clinical improvement of PD cardinal symptoms in the short-term, (ii) the “wearing-off” phenomenon [12,13], (iii) motor fluctuations and dyskinesia [10,14], (iv) systemic complications (gastrointestinal disorders, cardiovascular problems, hormonal dysregulation) [18,19], and (v) neuropsychiatric disorders (depression, anxiety, toxic psychosis) [11,18].
Atremorine is an option to minimize the “wearing-off” phenomenon,
extending the therapeutic effect of conventional antiparkinsonian drugs, and
reducing potential side effects, since the co-administration of Atremorine with
other antiparkinsonian drugs allows a dose reduction of conventional drugs by
25-50% with enhancement of clinical benefits and reduction of short- and
long-term adverse drug reactions.
However, although the dopaminergic surge induced by Atremorine is
proportional to basal DA levels in U and T PD patients, with a potential
200-500-fold increase over basal levels, its real potency and pharmacodynamic
and pharmacokinetic properties are highly influenced by genetic and
pharmacogenetic factors (Table 5).
Genes involved in the pharmacogenetic network include pathogenic, mechanistic,
metabolic, transporter and pleiotropic genes [26,27], and all these genes are
under the influence of epigenetic modifications (DNA methylation,
histone/chromatin remodeling, mRNA regulation) [28-30]. In recent years novel
evidence has demonstrated the impact of pharmacogenetics on anti-PD drug
efficacy and safety [11,31-34] (Table 1).
In the particular case of L-DOPA, the ANKK1, BDNF, LRRK2, and PARK2 genes are pathogenic genes
potentially involved in its effects. The CCK, CCKAR, CCKBR, DRD1, DRD2,
DRD3, DRD4, DRD5, GRIN2A, GRIN2B, HCRT, HOMER1, LMO3, and OPRM1
genes are mechanistic genes whose products influence L-DOPA efficacy and
safety. L-DOPA is a substrate of enzymes encoded by the COMT, CYP1A2,
CYP2B6, CYP2C19, CYP2D6, CYP3A4, CYP3A5, DBH, DDC, G6PD, MAOB, TH, UGT1A1, and
UGT1A9 genes responsible for its metabolism. SLC6A3 is the major
transporter of L-DOPA; and ACE, ACHE and APOE are pleiotropic
players in L-DOPA efficacy and safety [11] (Table 1). ADORA2A
SNPs and HOMER1 variants may be associated with L-DOPA-induced
dyskinesia and psychotic symptoms [35,36]. A haplotype integrating -141CIns/Del, rs2283265, rs1076560,
C957T, TaqIA and rs2734849 polymorphisms at the DRD2/ANKK1 gene region might also be associated with L-DOPA-induced
motor dysfunction [37]. SLC6A3 is a
genetic modifier of the treatment response to L-DOPA in PD [38].
Our
results illustrate the differential effect of APOE variants on
Atremorine-induced dopamine response (Figure
3-8, Table 5). APOE is a pleiotropic gene with enormous influence on
neurodegeneration, dementia and cerebrovascular disorders [39]. It has also
been extensively demonstrated that APOE-4 carriers are poor responders to
conventional drugs in dementia with and without a cerebrovascular component
[26,30,40-42]. In U PD patients, as previously mentioned, basal DA levels are
very homogeneous (<20 pg/mL) and Atremorine induces a spectacular increase
in DA levels (>2000 pg/mL in 80% of the cases), especially in APOE-2
carriers. The only U APOE-2/4 case, with a basal DA level of 12 pg/mL responded
with an increase in DA up to 7439pg/mL); and the only T APOE-2/4 case in our
sample, with a basal DA level of 21 pg/mL, showed a DA increase of 11912 pg/mL
one hour after Atremorine administration. According to our data, APOE-2
carriers are the best responders (Figure
6), APOE-3 carriers exhibit an intermediate response (Figure 7), and APOE-4 carriers show a moderate (significant)
response (Figure 8).
Similarly,
differential CYP-related Atremorine-induced dopamine responses have been
observed (Figure 9-12). L-DOPA is a
major substrate of CYP2D6, CYP2C19 and CYP3A4/5 enzymes [11] (Table 1). Assuming that the number of
cases included in this study is limited (and a larger sample is needed for
obtaining definitive conclusions), in general, CYP2D6-EMs are the best
responders, followed by CYP2D6-IMs; however, CYP2D6-PMs show a weaker response,
whereas CYP2D6-UMs exhibit an uneven response, with great heterogeneity and
response dispersion (Figure 9). In
an almost identical manner, CYP2C19-EMs are the best responders, CYP2C19-IMs
show an intermediate response (starting from higher basal DA values than EMs),
and CYP2C19-UMs show a weaker (significant) response than EMs and IMs, probably
due to a faster metabolization of L-DOPA (Figure
10). CYP2C9-IMs are better responders than EMs, and CYP2C9-PMs show a poor,
non-significant response (Figure 11).
Finally, CYP3A4/5-IMs are also better responders to Atremorine than
CYP3A3/4-EMs, though carriers of both geno-phenotypes are excellent responders,
and the few cases that harbor a CYP3A4/5-RM geno-phenotype show a weaker
(significant) response than EMs and IMs (Figure
12).
In
conclusion, Atremorine is a novel bioproduct derived from the Vicia faba pod with powerful
pro-dopaminergic properties in PD patients. The Atremorine-induced dopamine
response is genotype-dependent and is influenced by pleiotropic gene variants,
such as APOE, and CYP2D6, CYP2C19, CYP2C9 and CYP3A4/5 pheno-genotypes which
influence L-DOPA metabolism as well as other components present in the complex
composition of E-PodoFavalin-15999.
- von Campenhausen S,
Bornschein B, Wick R, Bötzel K, Sampaio C, et al. (2005) Prevalence and incidence of Parkinson's
disease in Europe. Eur Neuropsychopharmacol.15: 473-490.
- Zou YM, Liu J, Tian ZY, Lu D,
Zhou YY (2015) Systematic review
of the prevalence and incidence of Parkinson's disease in the People's
Republic of China. Neuropsychiatr Dis Treat 11: 1467-1472.
- Muangpaisan W, Hori H, Brayne
C (2009) Systematic review of the
prevalence and incidence of Parkinson's disease in Asia. J
Epidemiol 19: 281-293.
- Hirsch L, Jette N, Frolkis A,
Steeves T, Pringsheim T (2016) The
Incidence of Parkinson's Disease: A Systematic Review and Meta-Analysis. Neuroepidemiol
46: 292-300.
- Savica R, Grossardt BR, Bower
JH, Ahlskog JE, Rocca WA (2016) Time Trends in the Incidence of Parkinson Disease. JAMA
Neurol.
- Pringsheim T, Jette
N, Frolkis A, Steeves TD (2014)
The prevalence of Parkinson's disease: a systematic review and
meta-analysis. Mov Disord 29:1583-1590.
- Riedel O, Bitters D, Amann U,
Garbe E, Langner I (2016) Estimating
the prevalence of Parkinson's disease (PD) and proportions of patients
with associated dementia and depression among the older adults based on
secondary claims data. Int J Geriatr Psychiatry.
- Moisan F, Kab S, Mohamed F,
Canonico M, Le Guern M, et al.
(2015) Parkinson disease male-to-female ratios increase with age: French
nationwide study and meta-analysis. J Neurol Neurosurg Psychiatry.
- Miller DB, O’Callaghan JP
(2015) Biomarkers of Parkinson’s disease: Present and future. Metab
64:S40-S46.
- Katzenschlager R, Lees AJ
(2002) Treatment of Parkinson's
disease: levodopa as the first choice. J Neurol 249: II19-24.
- Cacabelos R (2012) World
Guide for Drug Use and Pharmacogenomics. Euro Espes Publishing, Corunna.
- Pahwa R, Lyons KE (2009) Levodopa-related wearing-off in
Parkinson's disease: identification and management. Curr Med Res
Opin 25: 841-849.
- Bhidayasiri R, Hattori N,
Jeon B, Chen RS, Lee MK, et al. (2015) Asian perspectives on the recognition and management of levodopa
'wearing-off' in Parkinson's disease. Expert Rev Neurother 15:
1285-1297.
- Haaxma CA, Horstink MW,
Zijlmans JC, Lemmens WA, Bloem BR, et al. (20015) Risk of Disabling Response Fluctuations and Dyskinesias for Dopamine
Agonists Versus Levodopa in Parkinson's Disease. J Parkinsons Dis 5:
847-853.
- Rascol O, Perez-Lloret S,
Ferreira JJ (2015) New treatments
for levodopa-induced motor complications. Mov Disord 30:1451-1460.
- Stowe R, Ives N, Clarke CE,
Deane K, van Hilten, et al. (2010) Evaluation of the efficacy and safety
of adjuvant treatment to levodopa therapy in Parkinson s disease patients
with motor complications. Cochrane Database Syst Rev 7: CD007166.
- Lertxundi
U, Isla A, Solinis MA, Domingo-Echaburu S, Hernandez R, et al. (2015) Anticholinergic burden in Parkinson's
disease in patients. Eur J Clin Pharmacol. 71: 1271-1277.
- Owolabi LF, Samaila AA,
Sunmonu T (2014) Gastrointestinal complications in newly diagnosed
Parkinson's disease: A case-control study. Trop Gastroenterol 35: 227-231.
- Tran T, Brophy JM, Suissa S,
Renoux C (2015) Risks of Cardiac
Valve Regurgitation and Heart Failure Associated with Ergot- and
Non-Ergot-Derived Dopamine Agonist Use in Patients with Parkinson's
Disease: A Systematic Review of Observational Studies. CNS Drugs
29: 985-998.
- Cacabelos R (2016) Bioactive
extract obtained from Viciafaba and its use in the treatment and/or
prevention of neurodegenerative diseases. European Patent EP16382138.
- Carrera I, Fernández-Novoa L,
Sampedro C, Aliev G, Cacabelos R (2016) Dopaminergic neuroprotection with
atremorine in Parkinson’s disease. Current Medicinal Chemistry.
- Boomsma
F, Alberts G, van Eijk L, Man in 't Veld AJ, Schalekamp MA (1993) Optimal
collection and storage conditions for catecholamine measurements in human
plasma and urine. Clin Chem 39: 2503-2508.
- Tuck
MK, Chan DW, Chia D, Godwin AK, Grizzle WE, et al. (2009) Standard
Operating Procedures for Serum and Plasma Collection: Early Detection
Research Network Consensus Statement Standard Operating Procedure
Integration Working Group. J Proteome Res 8: 113-117.
- Bouloux
P, Perrett D, Besser GM (1985) Methodological considerations in the
determination of plasma catecholamines by high-performance liquid
chromatography with electrochemical detection. Ann Clin Biochem 22:
194-203.
- Foti
A, Kimura S, DeQuattro V, Lee D (1987) Liquid-chromatographic measurement
of catecholamines and metabolites in plasma and urine. Clin Chem 33:
2209-2213.
- Cacabelos R, Cacabelos P,
Torrellas C, Tellado I, Carril JC (2014) Pharmacogenomics of Alzheimer's disease: novel therapeutic strategies
for drug development. Methods Mol Biol. 1175: 323-556.
- Cacabelos R, Torrellas C,
Carrera I (2015) Opportunities in Pharmacogenomics for the treatment of
Alzheimer’s Disease. Future Neurology 10: 229-252.
- Cacabelos R, Torrellas C
(2014) Epigenetic drug discovery
for Alzheimer's disease. Expert Opin Drug Discov 9: 1059-1086.
- Cacabelos R, Torrellas C
(2015) Epigenetics of aging and Alzheimer’s disease: Implications for
pharmacogenomics and drug response. Int J Mol Sci 16: 30483-30543.
- Cacabelos R, Torrellas C,
Teijido O, Carril JC (2016) Pharmacogenetic considerations in the
treatment of Alzheimer’s disease. Pharmacogenomics.
- Altmann V, Schumacher-Schuh
AF, Rieck M, Callegari-Jacques SM, Rieder CR, et al. (2016) Influence of genetic, biological and
pharmacological factors on levodopa dose in Parkinson's disease. Pharmacogenomics
17: 481-488.
- Jiménez-Jiménez FJ,
Alonso-Navarro H, García-Martín E, Agúndez JA (2016) Advances in
understanding genomic markers and pharmacogenetics of Parkinson's disease.
Expert Opin Drug Metab Toxicol 12: 433-448.
- Kurzawski M, Białecka M,
Droździk M (2015) Pharmacogenetic
considerations in the treatment of Parkinson's disease. Neurodegener
Dis Manag 5: 27-35.
- Schumacher-Schuh AF, Rieder
CR, Hutz MH (2014) Parkinson's disease
pharmacogenomics: new findings and perspectives. Pharmacogenomics
15: 1253-1271.
- Rieck M, Schumacher-Schuh AF,
Callegari-Jacques SM, Altmann V, Schneider Medeiros M, et al (2015) Is there a role for ADORA2A
polymorphisms in levodopa-induced dyskinesia in Parkinson's disease
patients?. Pharmacogenomics 16: 573-582.
- Schumacher-Schuh AF, Altmann
V, Rieck M, Tovo-Rodrigues L, Monte TL, et al. (2014) Association
of common genetic variants of HOMER1 gene with levodopa adverse effects in
Parkinson's disease patients. Pharmacogenomics J 14: 289-294.
- Rieck M, Schumacher-Schuh AF,
Altmann V, Francisconi CL, Fagundes PT, et al. (2012) DRD2 haplotype is associated with
dyskinesia induced by levodopa therapy in Parkinson's disease patients. Pharmacogenomics
13: 1701-1710.
- Moreau C, Meguig S, Corvol
JC, Labreuche J, Vasseur F, et al. (2015) Polymorphism of the dopamine transporter type 1 gene modifies the
treatment response in Parkinson's disease. Brain 138: 1271-1283.
- Cacabelos
R, Fernández-Novoa L, Lombardi V, Kubota Y, Takeda M (2005) Molecular
genetics of Alzheimer’s disease and aging. Meth Find Exp Clin Pharmacol 27: 1-573.
- Cacabelos R (2004) Genomic
characterization of Alzheimer’s disease and genotype-related phenotypic
analysis of biological markers in dementia. Pharmacogenomics 5: 1049-1105.
- Cacabelos R, Fernández-Novoa
L, Corzo L, Amado L, Pichel V, et al. (2004) Phenotypic profiles and
functional genomics in Alzheimer’s disease and in dementia with a vascular
component. Neurol Res 26: 459-480.
- Cacabelos R (2008)
Pharmacogenomics in Alzheimer’s disease. Meth Mol Biol 448: 213-357.
-
 Table 1
Table 1 -
Table 2
-
Table 3
-
Table 4
-
Table 5
-
Table 6
-
Table 7
-
Table 8
-
Table 9
-
Table 10
-
Table 11
-
Table 12




QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Advances in Nanomedicine and Nanotechnology Research (ISSN: 2688-5476)
- Journal of Genetics and Cell Biology (ISSN:2639-3360)
- Food and Nutrition-Current Research (ISSN:2638-1095)
- Journal of Microbiology and Microbial Infections (ISSN: 2689-7660)
- Journal of Veterinary and Marine Sciences (ISSN: 2689-7830)
- Proteomics and Bioinformatics (ISSN:2641-7561)
- Journal of Astronomy and Space Research


