3236
Views & Citations2236
Likes & Shares
Congenital poïkiloderma is infrequent and immunity
deficiency associated with poikiloderma is poorly reported. The association of
congenital poïkiloderma and neutropenia known as Clericuzio syndrome is rare.
The family involvement of this entity is exceptional. Analyzing two clinical
cases in a brother and sister, the authors underline the fact that the
cutaneous manifestations made possible to specify the etiology of this familial
congenital neutropenia after several years of diagnostic wandering.
Keywords:
Poïkiloderma, Neutropenia, Clericuzio, Syndrome, Black skin
INTRODUCTION
Congenital poïkiloderma is an infrequent dermatosis. The clinical
profile is very variable and the diagnosis is not always easy. It may associate
with mucous, phanerian and ocular involvement or integrate into complex
syndromes [1]. Immunity deficiency associated with poikiloderma is poorly
reported. The association of congenital poikiloderma and neutropenia known as
Clericuzio syndrome is rare [2]. It is a genodermatosis with autosomal
recessive transmission. The family involvements of this nosological entity are
exceptional. We report an observation in a brother and sister.
OBSERVATION
First Clinical Case
MK, 4 years old was born from non-consanguineous parents. She had
consulted at the age of 6 months in the department for a pruritic dermatosis.
She had recurrent cutaneous abscesses since the age of two months, associated
with pleuropulmonary and gastrointestinal infections. The cutaneous-mucous
examination found eczematous lesions of the limbs and trunk. The biological
balance showed hypochromic microcytic anemia, neutropenia at 250 cells / mm3.
The patient had been treated with topic steroïd and oral antibiotic therapy.
She then did not return to the appointments of controls for more than 3 years.
In font of recurrent infections, she reportedly had numerous consultations in
pediatrics where the various biological assessments carried out revealed a
permanent neutropenia. She was then treated with granulocyte growth factors. At
4 years old, we saw her again at the dermatological consultation. The clinical
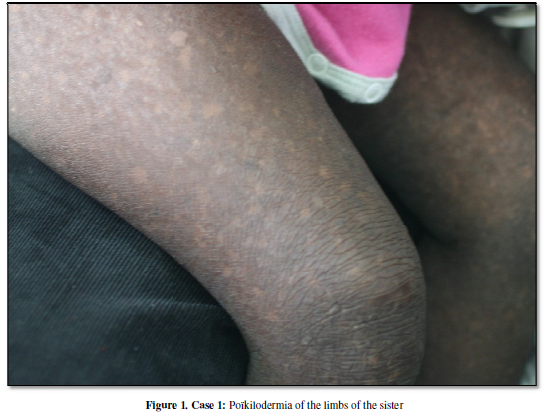
examination finds growth retardation associated with a diffuse poïkilodermia.
Indeed she had in most of the part of her body an hyperpigmentation associated
whith an hypopigmentation and a skin atrophy
( Figure 1). She had no
palmo-plantar keratoderma but a photosensitivity. The diagnosis of Clericuzio
syndrome was retained. There were no teeth abnormalities. The hairs and the
mucous membranes were normal. A topical steroid for the eczematous lesions and
a moisturizing cream were prescribed. The hematological disorder was follow up
by an hemato-pediatrician. The treatment leads to a stabilization of the
poikiloderma and the decreasing of the infections.
Second Clinical Case
MK's elder brother, 7 years old, was born at the end of a monofetal
pregnancy. He presented from the age of 2 months digestive disorders such as
gastroenteritis and repeated eczematous lesions. He had also been hospitalized
several times for multiple infections including bacterial pneumonia, urinary
tract infections and cutaneous abscesses.
The various biological examinations performed revealed a permanent
neutropenia ranging from 300 to 500 elements / mm3. At 6 months the child
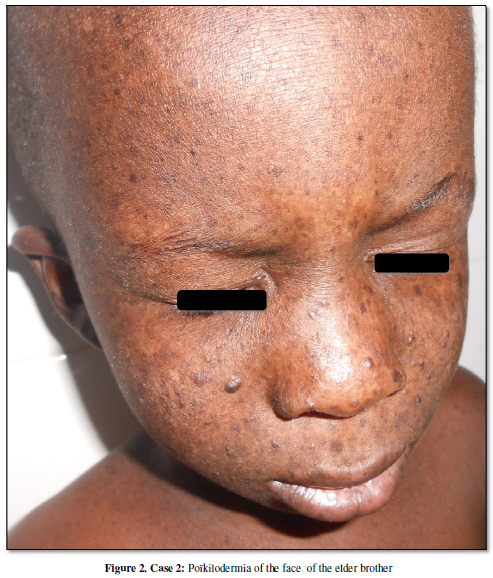
presented a progressive installation of poïkiloderma associated with plantar
hypopigmentation macules resembling Poikilodermie-Neutopenie syndrome of his
younger sister (Figure 2). A photosensitivity was noticed but he had no
palmo-plantar keratoderma. The hairs and
mucous membranes were also unremarkable. He had received the same treatment as
his sister with a stabilization of the lesions. No other relatives have similar
symptoms. Multidisciplinary management and follow up was recommended with
parental education in photoprotection, regular dermatological and hematological
monitoring.
DISCUSSION
Congenital poïkiloderma is rare. It may be the sign of some genodermatosis such as Rothmund-Thomson syndrome, Bloom syndrome, xerodermapigmentosum and congenital dyskeratosis [3]. Poikiloderma-Neutropenia syndrome was described for the first time in 1991 among the peoples from Navajo by Clericuzio. This syndrome is characterized by an eczematous onset of the skin, and secondarily by a poïkilodermal aspect of the skin, which predominates at the extremities with centripetal evolution [4]. It is frequently associated with recurrent bacterial and viral infections and hematological damage characterized by permanent neutropenia, leucopenia and thrombocytopenia. Patients often show a small size, pachyonychia and palmoplantar hyperkeratosis [5,6]. Bone abnormalities as well as malignant transformations have been reported [7]. This dominant autosomal genodermatosis is characterized by a mutation of the C16ORF 57 gene [8,9]. Management is difficult and consists essentially in the prevention of infections. A better understanding of genetic abnormalities will certainly lead to the development of targeted therapies. In this singular observation, the cutaneous manifestations made it possible to specify the etiology of this congenital neutropenia in a brother and sister after several years of diagnostic wandering.
CONCLUSION
Poikilodema-Neutropenia syndrome is a severe and rare genodermatosis.
This reported case shows that it is ubiquitous. In front of a congenital
poïkilodermal condition, the search of neutropenia and other signs of
Clericuzio syndrome is necessary in order to lead the patient early to a
multidisciplinary care.
- Farruggia
P, Indaco S, Dufour C, Lanza T, Mosa C, et al. (2014) Poikiloderma with
neutropenia: A case report and review of the literature. J Pediatr Hematol
Oncol 36: 297-300.
- Saussine A,
Petit A, Viguier M, Fremont G, Blanchet-Bardon, et al. (2012) Syndrome de
Clericuzio (syndrome « poïkilodermie-neutropénie »): une cause
exceptionnelle de poïkilodermie. Ann Dermatol Vénéréol 139: 181
- Van Hove JL,
Jaeken J, Proesmans M,
Boeck KD, Minner K, Matthijs
G, et al. (2005) Clericuzio type poikiloderma with neutropenia is distinct
from Rothmund-Thomson syndrome. Am J Med Genet A 132 : 152-158.
- Mostefai R,
Morice-Picard F, Boralevi F, Sautarel M, Lacombe D, et al. (2008)
Poikiloderma with neutropenia, clericuzio type, in a family from Morocco.
Am J Med Genet Part A 146A: 2762-2769.
- Chantorn R,
Shwayder T (2012) Poikiloderma with neutropenia: report of three cases
including one with calcinosis cutis. Pediatr Dermatol 29: 463-472.
- Mason PJ, Bessler M (2013) Poikiloderma with
neutropenia: beginning at the end. Blood 121: 872-874.
- Rodgers W,
Ancliff P, Ponting CP, Sanchez-Pulido L, Burns S, et al. (2013) Squamous
cell carcinoma in a child with Clericuzio-type poikiloderma with neutropenia. Br J Dermatol 168:
665-667.
- Concolino
D, Roversi G, Muzzi,GL, Sestito S, Colombo E, et al. (2010)
Clericuzio-Type PoikilodermaWith Neutropenia Syndrome in Three Sibs with
Mutations in the C16orf57 Gene: Delineation of the Phenotype. Am J Med
Genet Part A 152A: 2588-259.
- Turkan
Patiroglu H (2015) HalukAkar Clericuzio-type Poikiloderma with Neutropenia
Syndrome in a Turkish Family: a Three Report of Siblings with Mutation in
the C16orf57 gene. Iranian J Allergy, Asthma Immunology 14: 331-337.
QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Immunology Research and Therapy (ISSN:2472-727X)
- Journal of Cell Signaling & Damage-Associated Molecular Patterns
- International Journal of Clinical Case Studies and Reports (ISSN:2641-5771)
- Journal of Cardiology and Diagnostics Research (ISSN:2639-4634)
- International Journal of Anaesthesia and Research (ISSN:2641-399X)
- Oncology Clinics and Research (ISSN: 2643-055X)
- Journal of Spine Diseases


