1248
Views & Citations248
Likes & Shares
Immune
cell‐derived cytotoxic pathways have been implicated in antitumor immune
responses. The goal of this study is to characterize how these cytotoxic
pathways influence ovarian cancer development. We have utilized the TgMISIIR‐TAg transgenic mouse model which expresses the transforming SV40 TAg in
the ovary, leading to spontaneous development of ovarian tumors that closely
mimic human epithelial ovarian cancer. To test how perforin (Prf1), granzyme B
(GzmB) and interferon‐gamma (IFNg) impact tumor
occurrence and progression, we bred the TgMISIIR‐TAg
transgene into Prf1–/–, GzmB–/–, and IFNgR1–/–
mice. The transgenic females developed peritoneal tumors at 9‐15 weeks and succumbed at 184 ± 37 days of age with 100% penetrance
(n=41). Knockout of these cytotoxic genes does not affect tumor occurrence.
However, loss of function in the IFNg signaling pathway significantly expedited
tumor progression with all of the IFNg R1–/– TgMISIIR‐TAg females succumbing to tumor outgrowth at 167 ± 27 days of age
(p=0.0074, n=24). In contrast, loss of function of Prf1 or GzmB did not
significantly impact tumor progression and host survival. Since tumor cells in
the IFNg R1–/– TgMISIIR‐TAg
mice are IFNg R1 deficient, we used the implantable MOSEC (mouse
ovarian surface epithelial cell) tumor line to validate that IFNg R
signaling in host immune cells but not in tumor cells impacts tumor
progression. Indeed, when the IFNg ‐responsive MOSEC cells were
inoculated, IFNg R1–/– mice exhibited significantly higher
tumor burden compared to WT mice. Furthermore, a MOSEC‐splenocyte
co‐culture system confirmed that IFNg R1–/– immune cells
were less effective than WT immune cells in controlling MOSEC tumor growth in
vitro. Together, these results indicate that the IFNg R signaling
pathway plays an important role in restraining murine ovarian tumor
progression.
Keywords: Cytotoxic pathways, Interferon‐gamma (IFNg), Perforin (Prf1), Granzyme B (GzmB), Tumor immunity, Ovarian cancer.
INTRODUCTION
The intricacy
of immune activation versus immune suppression in the tumor environment affects
clinical outcomes. Several major immune cell‐derived
cytotoxic pathways have been shown to contribute to antitumor immune responses
[1-3]. For example, the perforin/granzyme pathway was believed to be critical
for immune surveillance against several types of blood cancers and epithelial
malignancies [4-7]. Perforin and granzymes were shown to be key effector
molecules for cytotoxic T cells and natural killer (NK) cells to eliminate
transformed tumor cells [8-11]. Interferon‐gamma (IFNg)
has also been shown to be broadly involved antitumor immune response mediated
by both T cells and innate immune cells [1,12]. Interestingly, these same
cytotoxic pathways have recently been implicated in suppressing antitumor
immune responses [13]. Several studies have revealed that perforin and granzyme
B can be used by regulatory T cells to suppress immune responses in mechanisms
involving damaging antigen presenting cells or effector lymphocytes
[14-17].
IFNg has also
been implicated in a complex network of immune suppression that involves
myeloid derived suppressor cells, regulatory T cells and indoleamine 2,3‐dioxygenase, [18-20] although IFNg
has the ability to directly inhibit tumor‐induced
regulatory T cell proliferation [21,22].
The opposite
impacts of suppressive immune cells versus antitumor immune cells have been
documented in both ovarian cancer patients and mouse models. While intratumoral
CD8+ T cell infiltration and a high CD8+/regulatory T cell ratio are associated
with favorable prognosis in ovarian cancer, accumulation of myeloid‐derived suppressor cells in the tumor environment
leads to weakened tumor immunity [23,24]. However, the cytotoxic molecular
pathways described above have not been carefully examined in ovarian cancer
models. Therefore, we have studied the roles of these cytotoxic pathways in
mouse models of ovarian cancer. We hypothesized that the perforin/granzyme and
IFNg pathways contribute to tumor immunity during ovarian tumor progression. We
have utilized a transgenic ovarian cancer model developed by Dr. Denise
Connolly [25]. The TgMISIIR‐TAg transgenic
mice specifically express the transforming SV40 TAg in the ovary and epithelium
of the female reproductive tract, leading to spontaneous development of ovarian
tumors that closely mimic human epithelial ovarian cancer [25,26]. To test
whether perforin (Prf1), granzyme B (GzmB) and interferon‐gamma (IFNg) are involved in tumor development in
this model, we have bred the TgMISIIR‐TAg transgene
into Prf1–/–, GzmB–/–, and IFNg R1–/– mice
and monitored tumor occurrence, progression and mouse survival. Results from
this transgenic model indicate that loss of function in the IFNg signaling
pathway significantly expedited tumor progression while loss of function of
Prf1 or GzmB did not significantly impact tumor progression. In addition, we
have used the IFNg ‐responsive
MOSEC ovarian tumor cell line to demonstrate that IFN signaling pathway in the
host immune cells plays an important role in restraining ovarian tumor
progression.
MATERIALS AND
METHODS
Mouse colonies
and tumor cell line
The TgMISIIR‐TAg transgenic mice in the C57BL/6J strain were
obtained from Dr. Denise Connolly at Fox Chase Cancer Center [25]. Prf1–/– and GzmB–/– mice
in the C57BL/6J strain have been generated and maintained as previously
described [9,15,27]. Wild‐type (WT) and IFNg
R1–/– mice in the C57BL/6J strain were purchased from the
Jackson laboratory. MOSEC tumor cell line was obtained from Dr. Kunle Odunsi at
Roswell Park Cancer Institute. MOSEC cells were transduced with a retroviral
vector to express luciferase and used for bioluminescence imaging to measure
tumor burden as previously described [15,21,27,28]. All mice were maintained in
SPF housing, and all experiments were conducted in accordance with the animal
care guidelines at Roswell Park Cancer Institute, using protocols approved by
animal studies committee.
Transgene
breeding, genotyping and monitoring tumor development
The TgMISIIR‐TAg transgenic mice were crossed with the Prf1–/–, GzmB–/–,
and IFNg R1–/– mice respectively to breed the
TgMISIIR‐TAg transgene into the Prf1–/–, GzmB–/–,
and IFNG R1–/– colonies. Presence of the transgene
was confirmed by PCR amplification of a 773‐bp fragment of
the large TAg using the TAg F4 forward primer (5′‐TGCATGGTGTACAACATTCC) and the TAg R1 reverse primer
(5′‐TTGGGACTGTGAATCAATGCC) as previously described
[25]. Prf1 locus was genotyped by PCR with a forward primer (5′‐TGGTCTGGTGGACTACAGCCTGGA) and a reverse primer (5′‐CCTGAACTCCTGGCCACCAAAGA), which produces a 300bp
fragment for WT allele and a 1500bp fragment for the knockout allele. GzmB
locus was genotyped by PCR with a forward primer (5′‐ACACAAGTACTCAGAAGACGTCA)
and reverse primer (5′‐TGAACACTGGGGAACCACT),
which produces a 690 bp fragment for WT allele and a 2.2 kb fragment for the
knockout allele. IFNG R1 locus was genotyped by separate PCR
with a common forward primer (5′‐TTG TTT GAT CCA
TTC TTT AAA TTG) paired with a reverse primer (5′‐GCT TCT TTG AAG
GGC TGG A) that produces a 310bp fragment for WT allele, and paired with
another reverse primer (5′‐AAT GGA GGG AGC
ACA GTT TG) that produces and a 450bp fragment for the knockout allele. The
resultant four genotypes of TgMISIIR‐TAg, Prf1–/– TgMISIIR‐TAg, GzmB–/– TgMISIIR‐TAg, and IFNg R1–/– TgMISIIR‐TAg female mice were evaluated weekly for tumor
development. After 12 weeks of age, all transgenic females were
monitored daily for intraperitoneal tumor formation and accumulation of ascites
fluid (detected by abdominal swelling). Time of death was recorded when tumor‐bearing mice that became moribund were sacrificed.
Bioluminescence
imaging in vivo and in vitro
WT and IFNg
R1–/– mice were injected intraperitoneally with 2.5 x 106 luciferase‐expressing MOSEC tumor cells. Bioluminescence imaging was performed to monitor
tumor burden in vivo as previously described [15,21,27,28]. To measure MOSEC tumor growth in
vitro, various doses of MOSEC cells
were cultured in 48‐well plates with a total volume of 1ml media for 92
hours, and then 20 µl D‐Luciferin
(15mg/ml) was added into each well, and bioluminescence imaging was performed
to measure tumor burden in each well. Tumor burden was expressed as photon flux (photons/sec).
MOSEC tumor and
splenocyte co‐culture
2 x 106 spleen
cells isolated from WT and IFNg R1–/– mice were
mixed with various doses (4000, 2000, and 1000) of luciferase‐expressing MOSEC cells respectively, and co‐cultured in 48‐well plates
with a total volume of 1ml media in each well. 92 hours later, 20 µl D‐Luciferin (15mg/ml) was added into each well, and
bioluminescence imaging was performed to measure tumor burden in each well as
described above.
RESULTS
Global
deficiency of IFNg R1, but not Prf1 or GzmB, significantly expedites TgMISIIR‐TAg‐driven tumor
progression.
The TgMISIIR‐TAg transgenic mice specifically express the
transforming SV40 TAg in the ovary and epithelium of the female reproductive
tract, leading to spontaneous development of ovarian tumors [25,26]. To test
whether perforin (Prf1), granzyme B (GzmB) and interferon‐gamma (IFNg ) are involved in controlling tumor
development driven by the TgMISIIR‐TAg transgene,
we have bred the TgMISIIR‐TAg transgene
into Prf1–/–, GzmB–/–, and IFN
R1–/– mice and monitored tumor development and mouse survival.
The transgenic females developed peritoneal tumors at 9‐15 weeks and
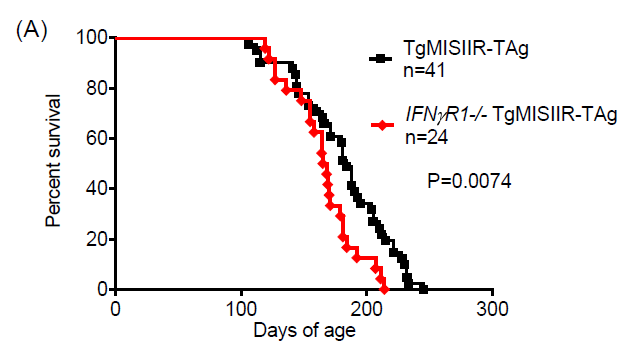
succumbed at 184 ± 37 days of age with 100% penetrance (n=41). Loss of function
in the IFN signaling pathway significantly expedited tumor progression
with all of th IFNg R1–/– transgenic females
succumbing to tumor outgrowth at 167 ± 27 days of age (p=0.0074,
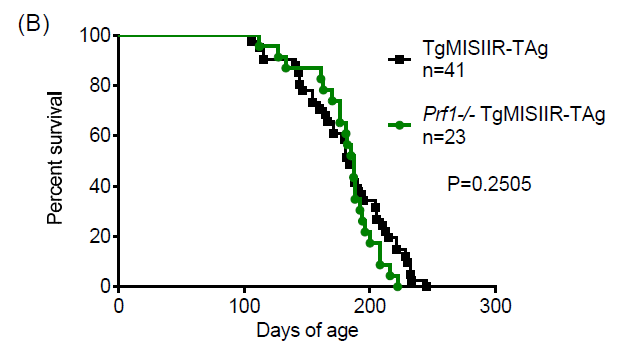
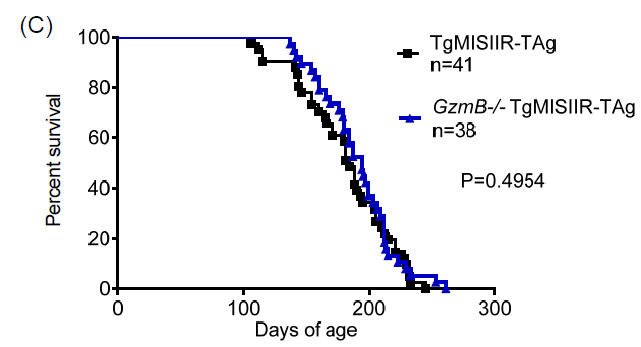
n=24) (Figure 1A). In contrast, loss of function of Prf1 or GzmB did not
significantly impact tumor progression as the Prf1–/– transgenic
females succumbed to tumor outgrowth at 187±27 days of age (p=0.2505, n=23) and
the GzmB–/– transgenic females succumbed at 191 ±
29 days (p=0.4954, n=38) (Figure 1B‐1C). It is clear that global loss of function of
these cytotoxic pathways does not affect tumor occurrence driven by the TgMISIIR‐TAg transgene, as 100% of the mice expressing the
transgene developed ovarian tumor in all four genotypes of mice. However, these
results highlight the importance of the IFNg R signaling pathway for limiting
tumor progression and prolonging survival of the tumor‐bearing mice.
It is important
to note that in the IFNg R1–/– TgMISIIR‐TAg mice described above, both the immune cells and
the SV40 TAg transformed tumor cells are IFNg R1–/–. Since IFNg
R1–/– mice can still produce IFNg , it is
therefore likely that loss of response to IFN by the IFNg R1–/– ovarian
tumor cells or by the IFNg R1–/– immune cells
accounts for the accelerated tumor progression. To test a hypothesis that loss
of function of the IFNg R signaling pathway in immune cells may dampen
antitumor response against the ovarian tumor cells, we employed the established
MOSEC ovarian tumor cell line, which was transformed through repeated passages
of mouse ovarian surface epithelium cells in vitro [29]. The
transformed MOSEC tumor cells were able to develop into peritoneal ovarian
tumors after inoculation into syngeneic C57BL/6 mice [29]. We first confirmed
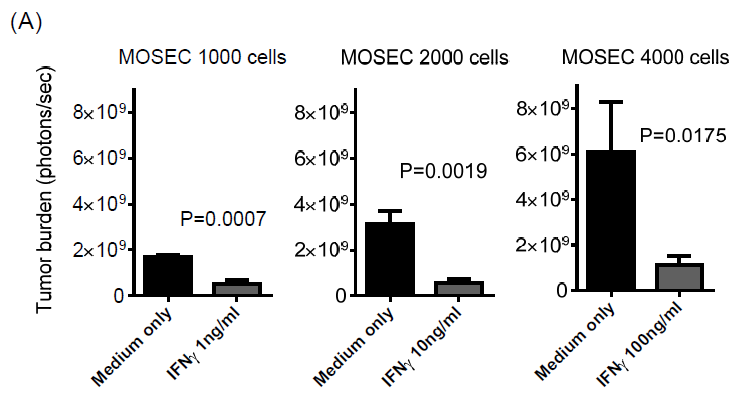
that the MOSEC tumor cells were able to directly respond to IFNg. Various doses
of recombinant IFN protein added to cell culture media were able to
suppress MOSEC cell proliferation consistently (Figure 2A), indicating
that MOSEC cells have a functional IFNg R signaling pathway. We also tried to
measure potential tumor cell killing by IFN treatment. However, no significant cell
death was observed by the methods of trypan blue, annexin V and 7AAD staining
(data not shown). Even with IFN treatment, tumor cells were still
proliferating, but at slower rates compared the non‐treated control
culture (Figure 2A). These results indicate that IFNg treatment
inhibits MOSEC tumor cell growth without causing substantial cell death.
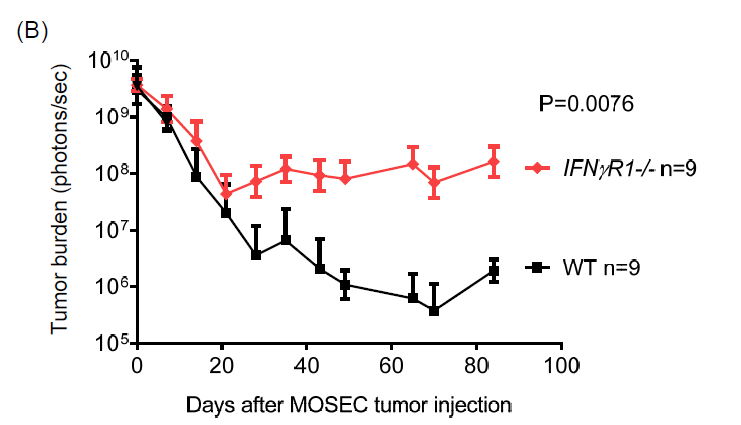
Next, to
investigate the effect of IFNg R signaling pathway in the host mice on ovarian
tumor progression, we inoculated IFNg R1–/– and WT
C57BL/6 mice with luciferase‐expressing
MOSEC tumor cells. Using bioluminescence imaging to serially monitor tumor
burden in vivo, we have observed significantly increased tumor
growth in IFNg R1–/– mice compared to that in WT
mice (Figure 2B). These results with the MOSEC tumor model are
consistent with the finding with the TgMISIIR‐TAg transgenic
model. Furthermore, these data indicate that IFNg R signaling in the host,
presumably in the host immune system, plays an important role in limiting
ovarian tumor growth.
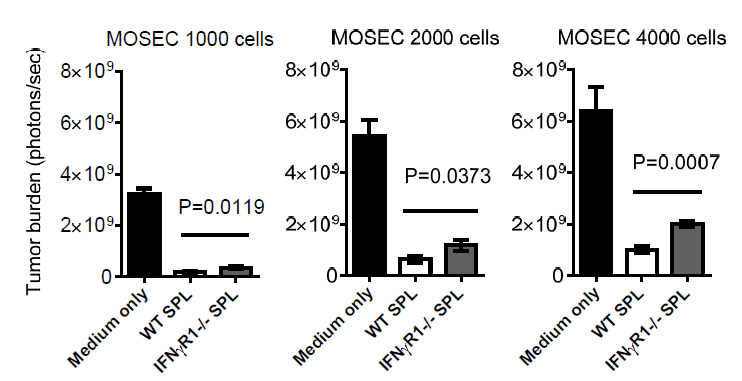
IFNg R1–/– spleen cells have diminished ability to control
MOSEC tumor growth in vitro compared to WT spleen cells
To further
define whether IFNg R signaling in the host immune system is responsible for
controlling ovarian tumor growth, we isolated spleen cells from WT and IFNg
R1–/– mice and tested their ability to control tumor growth in
vitro. As expected, spleen cells added to the MOSEC cell culture were able
to inhibit tumor cell growth (Figure 3). Furthermore, IFNg R1–/– spleen
cells showed reduced ability to control MOSEC tumor growth compared to WT
spleen cells. This in vitro co‐culture system
confirms that the IFNg R signaling pathway in the host immune cells is
important for an effective antitumor response against ovarian tumor cells.
DISCUSSION
It remains
challenging to diagnose ovarian cancer at early stages of disease because of
lack of symptoms and an effective screening system. Due to the often late
diagnosis at advanced stages of disease and limited efficacy of surgical and
chemotherapeutic strategies, ovarian cancer remains the most frequent cause of
death from gynecologic malignancy [29]. As promising new treatment modalities,
cancer immunotherapy strategies based on cancer vaccines and adoptive cellular
transfer are being actively investigated. This study aims to provide a better
understanding of how major cytotoxic molecular pathways in the immune system
impact ovarian cancer development and progression, which will be useful for
developing effective immunotherapy. We have provided solid evidence to show
that while global deficiency of GzmB, Prf1, or IFNg R does not
affect ovarian tumor occurrence driven by the aggressively transforming SV40
TAg, global loss of function of the IFNg R signaling pathway, but not Prf1
or GzmB, significantly accelerates tumor progression.
However, we
believe that this information has to be considered with caution because it has
recently been revealed that these cytotoxic pathways can be employed not only
by effector lymphocytes engaged in antitumor immune responses but also by
immune suppressor cells involved in dampening antitumor responses. For example,
global deficiency of GzmB or Prf1 could
disable their function in not only effector lymphocytes but also regulatory T
cells [14-17], resulting in opposite effects that could neutralize the overall
impact on tumor development. Likewise, global deficiency in the IFNg R signaling
pathway could have even broader effects because almost all innate and adaptive
immune cell types express functional IFNg receptor [18-22].
Although global
deficiency of the IFNg R signaling pathway could impact the functions of
effector immune cells as well as suppressor immune cells, this work with
ovarian cancer models shows that the oval impact of the IFNg R signaling
pathway has a positive effect on antitumor immune responses. In line with our
work, a previous study of ovarian cancer patients shows that loss of IFNg R in
ovarian tumor cells independently predicts poor prognosis [30], which may help
explain our observation since the tumor cells in our SV40‐driven spontaneous model are also deficient for
IFNg R. However, our study with the IFNg ‐responsive
MOSEC model presents a similar phenotype, suggesting that IFN R deficiency in
host immune cells is at least partially responsible for accelerated tumor
progression. Our previous study reveals that the numbers of mature CD4+,
CD8+ T cells and CD4+Foxp3+ Treg
cells are not altered in naive IFN R–/– versus WT mice.
However, tumor inoculation induces a higher expansion of Treg cells in IFNg R–/– versus
WT mice due to IFNg ‐mediated
inhibition of Treg cell expansion [21], which may partially contribute to the
increased tumor burden in the IFNg R–/– mice. In
contrast to these studies that describe a positive impact of IFNg on tumor
immunity, a recent study with murine melanoma model shows that IFNg
actually inhibits peptide vaccine‐induced tumor
immunity by inducing expression of high levels of noncognate MHC‐I and PD‐L1 molecules on
tumor cells [31]. While the discrepancy may result from many variables
including the different tumor types and distinct immunogenicity of the
different tumor models, we must acknowledge that IFNg signaling may play more
complex roles than simply promoting antitumor immune responses.
In summary, due
to the complex functions of these cytotoxic pathways in multiple immune cell
compartments that may have differential and even directly opposite impacts on
tumor immunity, it will be necessary to develop compartment‐specific deficiency of these cytotoxic pathways.
Further investigation with such improved models will provide deeper mechanistic
understanding of how these molecular pathways mediate interactions between
individual immune cell types and cancer cells, which may reveal novel targets
that can be manipulated to improve the safety and efficacy of immune‐based cancer therapy.
DISCLOSURE OF
CONFLICT OF INTEREST
The authors
have no potential conflict of interest to disclose.
ACKNOWLEDGEMENT
This work was
supported by a Young Investigator Development Award from Roswell Park Alliance
Foundation (X.C.) and a Pilot Study Award from Marsha Rivkin
Center for Ovarian Cancer Research (X.C.). N.D.L. was supported by a T32 pre‐doctoral training grant from NIH (CA085183).
1.
Dunn
GP, Koebel CM, Schreiber RD (2006) Interferons, immunity and cancer
immunoediting. Nat Rev Immunol
6: 836‐848.
2.
Lieberman
J (2003) The ABCs of granule‐mediated cytotoxicity: new weapons
in the arsenal. Nat Rev Immunol
3: 361‐370.
3.
Russell
JH, Ley TJ (2002) Lymphocyte‐mediated cytotoxicity. Annu Rev Immunol 20: 323‐ 370.
4.
Bolitho
P, Street SE, Westwood JA, et al. (2009) Perforin‐mediated
suppression of B‐cell lymphoma. Proc Natl Acad Sci USA106: 2723‐2728.
5.
Smyth
MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, et al. (2000) Perforin‐mediated cytotoxicity is critical for
surveillance of spontaneous lymphoma. J
Exp Med 192: 755‐760.
6.
Pardo
J, Balkow S, Anel A, Simon MM (2002) Granzymes are essential for natural killer
cell‐ mediated and perf‐facilitated
tumor control. Eur J Immunol 32:
2881‐2887.
7.
Leigh
ND, Bian G, Ding X, et al. (2014) A flagellin‐derived
toll‐like receptor 5 agonist stimulates cytotoxic
lymphocyte‐mediated tumor immunity. PLoS One 9: e85587.
8.
Cai
SF, Fehniger TA, Cao X, et al. (2009) Differential expression of granzyme B and
C in murine cytotoxic lymphocytes. J
Immunol 182: 6287‐6297.
9.
Fehniger
TA, Cai SF, Cao X, et al. (2007) Acquisition of murine NK cell cytotoxicity
requires the translation of a pre‐existing pool of granzyme B and
perforin mRNAs. Immunity 26: 798‐811.
10.
Kagi
D, Ledermann B, Burki K, et al. (1994) Cytotoxicity mediated by T cells and
natural killer cells is greatly impaired in perforin‐deficient
mice. Nature 369: 31-37.
11.
Revell
PA, Grossman WJ, Thomas DA, et al. (2005) Granzyme B and the downstream
granzymes C and/or F are important for cytotoxic lymphocyte functions. J Immunol 174: 2124‐2131.
12.
Street
SE, Trapani JA, MacGregor D, Smyth MJ (2002) Suppression of lymphoma and
epithelial malignancies effected by interferon gamma. J Exp Med 196: 129‐134.
13.
Cao
X (2010) Regulatory T cells and immune tolerance to tumors. Immunol Res 46: 79‐93.
14.
Boissonnas
A, Scholer‐Dahirel A, Simon‐Blancal V, et al. (2010) Foxp3+ T cells
induce perforin‐ dependent dendritic cell death in
tumor‐draining lymph nodes. Immunity 32: 266‐278.
15.
Cao
X, Cai SF, Fehniger TA, et al. (2007) Granzyme B and perforin are important for
regulatory T cell‐mediated suppression of tumor
clearance. Immunity 27: 635‐646.
16.
Gondek
DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ (2005) Cutting edge: contact‐mediated suppression by CD4+CD25+ regulatory
cells involves a granzyme B‐dependent, perforin‐ independent mechanism. J Immunol 174: 1783‐1786.
17.
Shevach
EM, DiPaolo RA, Andersson J, Zhao DM, Stephens GL, et al. (2006) The lifestyle
of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev 212: 60‐73.
18.
Gu
T, Rowswell‐Turner RB, Kilinc MO, Egilmez NK
(2010) Central role of IFNgamma‐ indoleamine 2,3‐dioxygenase axis in regulation of interleukin‐12‐mediated antitumor immunity. Cancer Res 70: 129‐138.
19.
Katz
JB, Muller AJ, Prendergast GC (2008) Indoleamine 2,3‐dioxygenase
in T‐cell tolerance and tumoral immune escape. Immunol Rev 222: 206‐221.
20.
Medina‐Echeverz J, Haile LA, Zhao F, et al. (2014)
IFN‐gamma regulates survival and function of
tumor‐induced CD11b+ Gr‐1high
myeloid derived suppressor cells by modulating the anti‐apoptotic
molecule Bcl2a1. Eur J Immunol
44: 2457‐2467.
21.
Cao
X, Leonard K, Collins LI, et al. (2009) Interleukin 12 stimulates IFN‐gamma‐mediated
inhibition of tumor‐induced regulatory T‐cell proliferation and enhances tumor
clearance. Cancer Res 69: 8700‐8709.
22.
Kilinc
MO, Aulakh KS, Nair RE, et al. (2006) Reversing tumor immune suppression with
intratumoral IL‐12: activation of tumor‐associated T effector/memory cells, induction
of T suppressor apoptosis, and infiltration of CD8+ T effectors. J Immunol 177: 6962‐6973.
23.
Khan
AN, Kolomeyevskaya N, Singel KL, et al. (2015) Targeting myeloid cells in the
tumor microenvironment enhances vaccine efficacy in murine epithelial ovarian
cancer. Oncotarget 6: 11310‐11326.
24.
Sato
E, Olson SH, Ahn J, et al. (2005) Intraepithelial CD8+ tumor‐infiltrating lymphocytes and a high
CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian
cancer. Proc Natl Acad Sci USA 102:
18538‐18543.
25.
Connolly
DC, Bao R, Nikitin AY, et al. (2003) Female mice chimeric for expression of the
simian virus 40 TAg under control of the MISIIR promoter develop epithelial
ovarian cancer. Cancer Res 63:
1389‐1397.
26.
Hensley
H, Quinn BA, Wolf RL, et al. (2007) Magnetic resonance imaging for detection
and determination of tumor volume in a genetically engineered mouse model of
ovarian cancer. Cancer Biol Ther
6: 1717‐1725.
27.
Bian
G, Ding X, Leigh ND, et al. (2013) Granzyme B‐Mediated
Damage of CD8+ T Cells Impairs Graft‐versus‐Tumor
Effect. J Immunol 190: 1341‐1350.
28.
Ding
X, Bian G, Leigh ND, et al. (2012) A TLR5 agonist enhances CD8(+) T cell‐mediated graft‐
versus‐tumor effect without exacerbating
graft‐versus‐host
disease. J Immunol 189: 4719‐4727.
29.
Roby
KF, Taylor CC, Sweetwood JP, et al. (2000) Development of a syngeneic mouse
model for events related to ovarian cancer. Carcinogenesis 21: 585‐591.
30. Duncan TJ, Rolland P, Deen S, et al. (2007) Loss of IFN gamma receptor is an independent prognostic factor in ovarian cancer. Clin Cancer Res 13: 4139‐4145.
31. Cho HI, Lee YR, Celis E (2011) Interferon gamma limits the effectiveness of melanoma peptide vaccines. Blood 117: 135‐144.
QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Dermatology Clinics and Research (ISSN:2380-5609)
- International Journal of AIDS (ISSN: 2644-3023)
- Journal of Clinical Trials and Research (ISSN:2637-7373)
- International Journal of Clinical Case Studies and Reports (ISSN:2641-5771)
- International Journal of Anaesthesia and Research (ISSN:2641-399X)
- Journal of Cardiology and Diagnostics Research (ISSN:2639-4634)
- Journal of Renal Transplantation Science (ISSN:2640-0847)



