15749
Views & Citations14749
Likes & Shares
Adoptive
T cell transfer (ACT) can mediate objective responses in patients with advanced
malignancies. There have been major advances in this field, including the
optimization of the ex vivo generation
of tumor-reactive lymphocytes to ample numbers for effective ACT therapy via
the use of natural and artificial antigen presenting cells (APCs). Herein we
review the basic properties of APCs and how they have been manufactured through
the years to augment vaccine and T cell-based cancer therapies. We then discuss
how these novel APCs impact the function and memory properties of T cells.
Finally, we propose new ways to synthesize aAPCs to augment the therapeutic
effectiveness of antitumor T cells for ACT therapy.
Keywords:
Adoptive cell transfer, Artificial antigen presenting cells (aAPCs), T
cells, Cancer immunology, Immunotherapy
INTRODUCTION
The
isolation, expansion and infusion of tumor-reactive T cells into patients,
called adoptive T cell transfer (ACT), can mediate objective responses in
individuals with late-stage tumors [1-3]. Since its development, there have
been several advances in this field, including 1) the optimal way to
precondition a patient with chemotherapy prior to infusing T cells and 2) how
to optimally generate sufficient numbers of T cells using unique cytokines,
small molecules and antigen presenting cells (APCs) to instill durable memory
responses to tumors. Herein, we focus on the impact of natural and artificial
aAPCs in shaping the biology of tumor reactive T cells. We then suggest
creative ways to synthesize aAPCs to enhance the persistence, cellular
bio-energetic, and antitumor capacity of transferred T cells in patients.
Adoptive T Cell
Transfer
Adoptive
T cell transfer (ACT) is a customized immunotherapy for patients with advanced
malignancies [1,4-6]. This approach involves rapid ex vivo expansion of autologous or allogeneic T cells to
significant numbers (10e9-11), followed by infusion into a
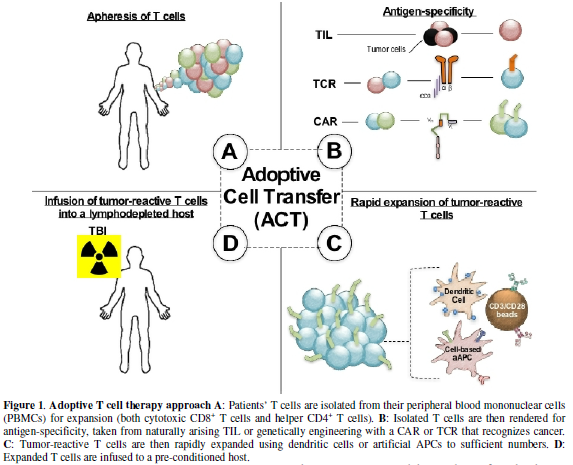
pre-conditioned individual, as shown in Figure
1. Also, detailed in Figure 1
are the different types of APCs that may be used to expand T cells ex vivo. These APCs include natural
dendritic cells (DCs) as well as artificial cell or bead based DCs. T cell
products can originate from the tumor (called tumor infiltrating lymphocytes,
TILs) or from the peripheral blood. Peripheral blood lymphocytes are rendered
antigen-specific by engineering expressions of T cell receptors (TCRs) or
chimeric antigen receptors (CARs). Autologous ACT is a promising treatment for
individuals with metastatic melanoma with complete response rates in 50% of TIL
therapies [3,7-9]. Allogeneic therapies, specifically CD19-CAR-specific
transfers, can render objective responses in 83% of patients with acute
lymphoblast leukemia (ALL) [5,10,11] and 27% of patients with chronic
lymphoblastic leukemia (CLL) [12-14]. A major advancement in adoptive
immunotherapy includes host preconditioning prior to cell transfer. The
mechanisms underlying the effects of lymphodepletion prior to ACT are discussed
below.
Lymphodepletion Enhances ACT Therapy
ACT
clinical trials in the 1990s infused tumor-specific TILs that yielded
disappointing responses in melanoma patients [15], mediating objective
responses in approximately 30% of patients. However, more than half of patients
with advanced melanoma achieved an objective response if they were first
preconditioned with a cyclophosphamide/ fludarabine lymphodepletion regimen
prior to adoptive transfer of TILs [16]. Importantly, some of these patients experienced
long-term curative responses with this approach. Finding that host
preconditioning augments the antitumor activity of transferred T cells has
advanced the field, thus promoting other investigators around the world to
perform this therapy in their patients [3,17]. Several mechanisms underlie how
lymphodepletion augments ACT therapy, including the elimination of host immune
cells that suppress infused TIL. These host cells include host regulatory T
cells (Tregs) [18,19] or other host lymphocytes that compete for homeostatic
cytokines, such as interleukins 7 and 15 (IL-7 and IL-15) [18,20].
Lymphodepletion also activates the innate immune system through gut microbes
that translocate from the injured bowel thereby augmenting the function and
persistence of infused T cells [21]. Finally, lymphodepletion ablates MDSCs and
regulatory B cells (Bregs) in the tumor microenvironment, which can impair the
antitumor activity of infused T cells. Thus, host preconditioning provides an
environment where the transferred lymphocytes can engraft and persist in the
patient.
Lymphodepletion
is not the only factor influencing clinical responses in patients treated with
ACT therapy. Emerging findings now show that the ability to expand T cells to
sufficient numbers without compromising their antitumor efficacy is a crucial component
for successful ACT trials. The importance of how cellular product is expanded and the ideal properties of a
therapeutic T cell are key concepts in adoptive immunotherapies. For example,
the differentiation status and cellular energetics of tumor-reactive
lymphocytes are important for sustaining their durability in the host [22-24].
Below we review recent reports that describe some of the ideal properties of T
cells that mediate the highest antitumor responses in vivo.
Central Memory T Cells in Antitumor Immunity
Lymphocytes
naturally progress through differentiation states, which are governed by
antigen stimulation from dendritic cells (DCs). It is becoming clearer that T
cell’s antitumor efficacy is denoted by the T cell’s differentiation state
[25-28]. Their naïve, stem, central and effector memory profile has long been
associated with their differentiation state, which can be characterized by the
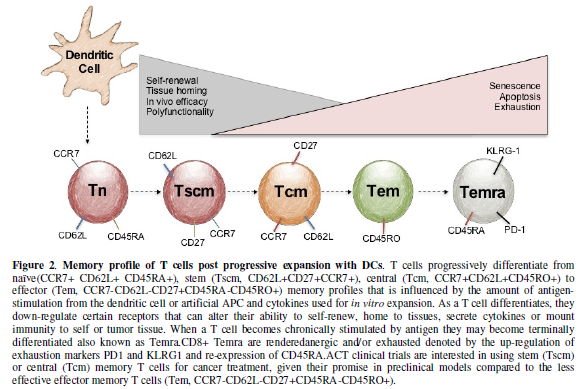
expression of certain surface receptors [25,29,30], as shown in Figure 2. Historically, T cells
selected for transfer possessed an effector memory phenotype (CD62L-CD45RA+
expression), with the ability to secrete IFNγ in vitro and have in vivo
cytolytic capacity [26]. Against dogma, Restifo, Gattinoni and co-workers
reported that less differentiated stem and central memory CD8+ T
cells, denoted by their expression of CD62L, CCR7 and β-catenin, were superior
at regressing tumors than effector memory CD8+ T cells in mice
[16,26]. This discovery resulted in part from the finding that tumor-specific
CD8+ central memory cells can persist longer in vivo than their CD8+ effector memory counterparts [16,22,31]. To
further investigate the robustness of central memory T cells, the Dirk Busch
lab conducted multiple serial transfer experiments where a mere 100 central
memory T cells and 100 effector memory T cells were infused into mice with an
infectious disease. They found that the central memory T cells cleared listeria
far better than the effector memory T cells [31]. Moreover, in a second and
third serial transfer experiment, 100 central memory T cells, but not the 100
effector memory T cells, continued to protect the animal from are-challenge of
listeria. Given the ability of ACT with less differentiated T cells to deliver
robust antitumor responses in mice, clinical trials are underway to use
enriched CD62L+T cells to treat patients with advanced malignancies
[32]. Designing an expansion protocol with natural or artificial antigen
presenting cells that specifically support the expansion of central over
effector memory CD8+ T cells might have profound implications for
next generation ACT clinical trials. For example, several investigators are
exploring the role of TCR “signal strength” improving or hindering the
antitumor efficacy of T cells with CD3/CD28 activator beads [33,34], with cell
culture plates adhered with anti-CD3 and soluble anti-CD28 [35], or mAbs of CD3
and CD28 [36]. It is becoming clearer that the length of time T cells are
initially activated with TCR stimulation, the progression of differentiation
occurs, which can negatively prime T cells in
vitro, decreasing cytokine production and hindering their ability to
regress tumor in vivo [33-35].
Another key concept about ex vivo T cell activation, are the co-stimulation of
CD28 enhancing progressive differentiation through up-regulating
glycolysis via the mTOR pathway [36]. The advantages of using aAPCs to
prime T cells include two things: 1. Using various costimulatory molecules,
other than CD28; like ICOS, to preferentially expand subsets of T cells that
will develop a higher antitumor efficacy [33] and 2. Manipulating the duration
of aAPCs to activate T cells in vitro
by length of duration in culture or the amount of beads placed in culture
[33,34].
APC Platforms for the ex vivo Expansion of T cells
The
development of affordable platforms to expand sufficient numbers of T cells
with potent antitumor activity has been a key goal in the field. Initial ex vivo T cell expansion protocols used autologous
dendritic cells (DCs) that, when co-cultured with T cells, preferentially
expanded TILs to treat patients with melanoma [37]. However, the ability to
generate enough of antigen-specific T cells with this approach varied between
patients, likely due to the fitness of the patient’s T cells and/or DCs
[38-41]. There are many reasons why autologous DCs can be challenging to work
with. For example, DC-based T cell expansions are complex, requiring multiple
cultures, numerous cytokines and extended times for cell expansion. Also, DCs
can possess a suppressive phenotype, which does not permit the generation of T
cells with a desired phenotype [39-41]. Ultimately these hurdles contribute to
complex protocols that are technically complex and costly to reproduce, thus
restricting TIL therapies to only a few institutes around the world. These limitations prompted the quest
for the generation of clinical grade artificial antigen presenting cells
(aAPCs) that could rapidly and simply expand tumor-reactive T cells.
In the
following sections, we discuss how natural DCs (Figure 3) augment TIL based immunotherapy for cancer. We then
focus on the evolution of aAPCs (Figure
4) through the years. We discuss immortalized K562 and paramagnetic aAPCs
and their role in tumor immunity. The potential of aAPCs is limitless: they can
be decorated with any number of co-stimulatory molecules to augment antitumor T
cells for ACT therapy.
Natural Versus Artificial APCs
Dr. Ralph
Steinman and his team discovered an APC called a DC in the 1970s and was
awarded a Nobel Prize in 2011 for this work [42]. DCs are composed of two
distinct lineages: the myeloid and plasmacytoid lineage [43-45]. Immature DCs
mature via distinct stimuli in a stepwise fashion. Immature DCs maintain
tolerance to self-antigens and blunt immunity to cancer via their expression of
various regulatory molecules (such as CTLA-4 or PD-1) and cytokines (i.e. IL-10
and TGF-). In contrast, mature DCs, activated in response to microbial signals
(toll-like receptor ligands), trigger strong effector T cell responses against
antigens [44,46]. It is known that DCs are phagocytic cells of the immune
system that degrade pathogens and can clear tumors by a process called
macropinocytosis [47]. The main role of mature DCs are to sense antigens and
produce mediators that activate other immune cells, particularly T cells [48].
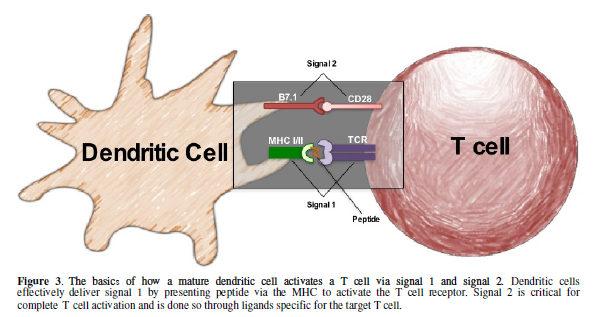
DCs are potent stimulators for lymphocyte activation as they express MHC molecules
that trigger TCRs (signal 1) and co-stimulatory molecules (signal 2) on T cells
[46]. This classic signal 1 signal 2 model: shown in Figure 3, illustrates how a mature DC can activate T cells via
TCR/MHC and B7.1/CD28 ligations [44,46].
Additionally, DCs also secrete cytokines that support T cell expansion;
many investigators call this signal 3 [49]. Unlike B-cells that can recognize
whole antigens, T cells require presented antigen in the form of a processed
peptide to recognize foreign pathogens or tumor [46]. Presentation of peptide
epitopes derived from pathogen/tumor proteins is achieved through specialized
cell-surface glycoproteins called major histocompatibility complex (MHC)
molecules. MHC class I (MHC-I) and MHC class II (MHC-II) molecules present
processed peptides to CD8+ T cells and CD4+ T cells,
respectively [46]. Importantly, DCs
home to inflammatory sites containing abundant T cell populations to foster an
immune response [44,50]. Thus, DCs can be a crucial component of any
immunotherapeutic approach [51], as they are intimately involved with the
activation of the adaptive immune response [45,51].
The
ability to generate DCs ex vivo from
blood monocytes has permitted immunologists to use them clinically as vaccines
or in ACT protocols to expand T cells. In the context of vaccines, DC therapy
can enhance T cell immune responses to a desired target in healthy volunteers
or patients with infectious disease or cancer [37,52]. Treatment with immature
DCs, in stark contrast, inhibits CD8+ T cell effector responses by
propagating regulatory T cells [53]. Thus, DC immunization is of interest to
the field of immunotherapy for cancer, infectious diseases and autoimmunity.
Dentritic Cells in ACT Clinical Trials
Several
current protocols for the expansion of tumor-specific T cells use autologous
DCs derived from patient’s PBMCs. Immature DCs are activated and matured with
the polarizing cytokines GMC-SF and IL-4 [37,52]. Once matured, they are pulsed
with a peptide of interest or lysed tumor cells. Mature/antigen-pulsed DCs are
then co-cultured with tumor-derived CD8+T cells where they undergo
weekly DC re-stimulation until enough TILs are expanded for the desired assay
or for therapeutic use [52]. A few clinical trials have successfully treated
melanoma patients with expanded TILs using this approach [37,54]. While this
therapy can be very effective in treating patients with melanoma, there exist
hurdles in this strategy in generating TILs of sufficient quality and quantity.
As stated earlier, the limitation of using patient derived-DCs for stimulation
and expansion of T cells is that the antitumor responses are not always
consistent across donors and that generation of effector memory T cells have
diminished function or persistence [39]. For ACT clinical trials, the
generation of DCs to reliably expand
TILs or CAR T cells are difficult and expensive. The culture process is resource
intensive and requires an expensive complex cytokine cocktail. Moreover, there
is variability among the donors DCs’ capacity to expand T cells ex vivo [40,41]. Perhaps most
concerning, DCs are often dysfunctional in patients with cancer [39-41].
Consequently, investigators have spent considerable time and money generating
various types of manufactured DCs called aAPCs to better expand T cells with
improved responses to antigen. We review some of these aAPCs directly below.
The K562 Approach: A Cell-based Artificial
aAPC
Translational
immunologists have successfully expanded human T cells with aAPCs instead of
natural APCs. One common approach is the use of the K562 cell line. K562 cells
do not express MHC molecules or co-inhibitory/co-stimulatory molecules,
therefore preventing allogeneic T cell responses. However, they do express
adhesion molecules (ICAM-1 and LFA-3) needed for effective T cell-APC
interactions [55,56]. Additionally, K562 cells secrete M-CSF, IL-6, IL-8,
TGF-β, and MIP-1α but do not secrete the γ-chain receptor cytokines IFNγ or
IL-10 [55]. All advantages aside, this original K562-based aAPC did not meet
GMP requirements for clinical use due to unstable transfection of surface
molecules that required continuous antibody selection [56]. To address this
limitation, several laboratories have improved this aAPC system by genetically
redirecting them with a lentiviral vector system to express an array of
co-stimulatory molecules and cytokines. The June laboratory generated
clinical-grade K562-cell–based aAPCs that could stably express 7 genes using
their lentiviral vector system [55,57]. These aAPCs mediated the expansion of
human T cells as effectively as natural DCs. These aAPCs were found to be
particularly effective at expanding human CD8+ T cells. Importantly,
the various co-stimulatory ligands engineered on this aAPC could expand
terminally differentiated CD28-CD8+ T cells without the
normal requirement of exogenous IL-2 used in nearly all cell culture processes
today. This K562-based aAPC has significant promise for designing next
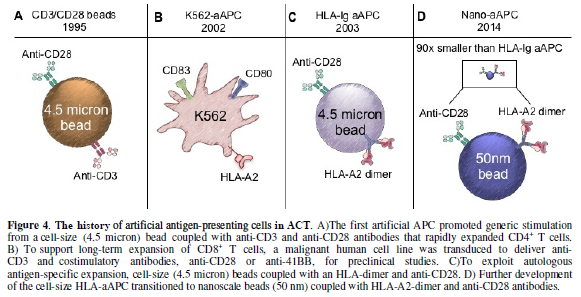
generation T cell-based cancer immunotherapies. As shown in Fig. 4B, a clinical grade and
GMP-quality K562-based aAPC-A2 line called clone 33 was used to expand MART-1
specific T cells against advanced melanoma [58]. These K562-aAPCs were
transfected with four non-retroviral plasmids that encode for HLA-A*02:01 (A2),
CD80, CD83, and a puromycin resistance gene (Figure 4B). In comparison to the natural DC expansion platform,
aAPC-A2 clone 33 similarly expanded MART-1-specific T cells from both healthy
donors and patients with metastatic melanoma (19-49% tetramer positive)
[58,59]. Clinical trials are beginning to use this aAPC in combination with
various treatment modalities, such as Ipilimumab [58]. However, the K562 aAPC
platform has not been widely used for cancer therapy, largely due the fact that
these cells are derived from a malignant clone. Although K562-aAPCs are
irradiated before co-cultured with T cells so that none of them are detected
after T cell expansions, there are appropriate reservations in infusing T cell
products with a malignant cell line into cancer patients.
Dynabeads for Expanding Polyclonal T Cells
To avoid ex vivo expansion of human T cells with
the K562-aAPC cell lines, ACT protocols have utilized a bead-based aAPC
approach for cell expansions. ACT clinical trials expand lymphocytes with
paramagnetic beads coated with CD3 and CD28 agonist antibodies (called
activator beads). Fig. 4A
illustrates the bead construct of simultaneously delivering both signal one
(anti-CD3) and signal two (anti-CD28) for non-specific stimulation that
mediates robust expansions of human T cells for up to several weeks [60,61].
This approach reproducibly drives multiple rounds of proliferation of T cells,
and can result in greater than 1 × 109-fold expansion of the input
cell population [62]. This large expansion is due, at least in part, to the
CD28-mediated induction of telomerase in CD4+ T cells [62,63]. Despite
extensive ex vivo replication,
bead-expanded T cells retain in vivo
proliferative capacity. Furthermore, it was discovered that these
anti-CD3/28-coated beads also promoted vigorous expansion of CD4+ T
cells from patients with HIV. Interestingly, during expansion the number of
HIV-positive CD4+ T cells declined overtime to nearly undetectable
levels [60]. This important discovery led to the manufacturing of GMP-compliant
anti-CD3/CD28 beads and the first Phase I clinical trial conducted by the June
and Riley lab at the University of Pennsylvania [61]. Since then,
anti-CD3/CD28-coated beads have been extensively used to expand T cells for use
in multiple clinical trials. For example, these beads are used to expand T
cells that are genetically redirected to express chimeric antigen receptors
that recognize CD19-postiive hematological malignancies (i.e. CD19-CARTs)
[5,64,65]. In Phase 1 clinical trials, patients receiving CD19-specific CAR
therapies have rendered outstanding objective response rates of 93% in ALL, 63%
in CLL, and 36% in lymphoma [5,6,67]. While these CD3/CD28 activator beads deliver
robust expansion of engineered tumor-reactive T cells, development of
antigen-specific expansion platforms to transfer autologous tumor-specific T
cells is a long-term goal within the field. Discussed below are novel
bead-based aAPCs that can rapidly expand antigen-specific T cells from healthy
donors.
Harnessing Antigen Specific Activation with
aAPCs
Besides
TIL stimulation with autologous dendritic cells, earlier attempts to generate
antigen-specific T cells with artificial APCs included either cell-based
approaches with the Drosophila spp.
cell line, the K562 cell line or exosomes coated with MHC class I peptides and
B7.1/2 (a natural ligand for CD28) molecules [56,57,68]. In 2003, Oelke and
colleagues developed a bead-based approach to expand antigen-specific T cells,
shown in Fig. 4C. This aAPC is a
magnetic bead of cell-size (4.5 micron) coated with HLA-A2-Ig dimer molecules
(signal 1) and anti-CD28 antibodies (signal 2) [69-71]. HLA-Ig aAPCs expanded
CMV- and MART-1-specific T cells 106-fold in less than two months
[69]. Additionally, bioluminescence technology revealed that MART-1 specific T
cells expanded with HLA-Ig-based aAPCs trafficked to the HLA-A2+ but not to
HLA-A2- melanoma tumors [72]. Furthermore, the tumor growth was inhibited,
confirming that these T cells eradicated tumor in an antigen specific manner
[72]. This technology progressed to a nanoscale platform, offering new
advantages in how immunologists expand antigen-specific T cells for cancer
therapies [73,74].
Nanoscale Expansion Platforms
for ACT
Recent
evidence suggests that nanosize-aAPCs (50 nm), which are 90-times smaller than
traditional CD3/CD28 beads (4.5 um) can be more advantageous at expanding T
cells ex vivo. First, these beads
mimic natural biology, as the initial TCR engagements on T cells occur at
nanoscale clusters that could enhance antigen-specific activation [73-76]. The
size of the nano-aAPCs may be able to select T cells with a low precursor
frequency in the tumor and blood [76,77]. The Oelke lab’s nanoscale aAPC
successfully expanded antigen-specific T cells ex vivo with high antitumor activity in vivo [74]. These nanoscale aAPCs are biocompatible iron-dextran
paramagnetic nanoparticles (50 nm) or are avidin-coated quantum dot
nanocrystals, (30 nm) [74]. Each type of nano-aAPC is coupled with MHC-Ig (or
HLA-Ig) dimers, Kb-Ig and Db-Ig (signal 1) and CD28
antibodies (or other costimulatory agonists) for signal 2: shown in Figure 4D. These nano-aAPCs were shown
to expand highly functional SIY-specific or gp100-specific T cells after
re-stimulation as well as mediate comparable Pmel (gp100-specific) expansions
to the micro-scale aAPCs [74]. Additionally, nano-aAPCs inhibited B16 melanoma
tumor growth in mice by expanding antigen-specific T cells with function and
persistence in vivo [74].
Importantly, this preclinical finding can be translated to human T cell assays,
as nano-aAPCs also mediated an 800-fold expansion of human T cells that could
recognize and lyse influenza [74].
To expand
rare antigen-specific precursors that lyse tumors, such as NY-ESO-1 and
WT-1-reactive T cells, novel enrichment and expansion (E+E) protocols have been
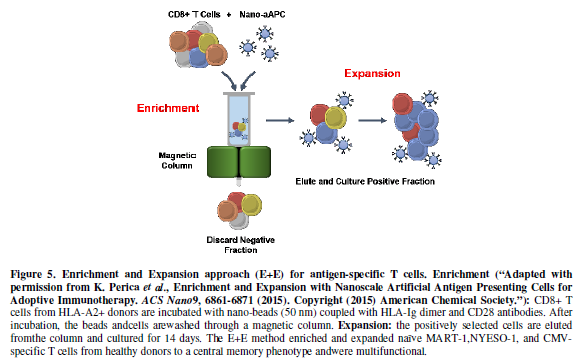
reported [73]. Figure 5 demonstrates
the E+E method, where first, antigen-specific CD8+ T cells from HLA-A2+ donors
are incubated with paramagnetic nanoparticles decorated with HLA-Ig-dimers
pulsed with MART-1 peptide (for example) and anti-CD28. This culture is then
enriched for antigen-specificity through a magnetic column, where positively
selected cells are cultured for 14 days [73]. This approach mediates robust
expansions for MART-1 and NYESO-1-specific T cells [73]. Additionally, this
novel aAPC platform can expand neoantigen-specific T cells using predicted
neo-epitopes obtained from a sequenced tumor [73]. This E+E platform could make a substantial contribution to next
generation ACT trials, where rare yet very effective T cells can be expanded
with a durable memory phenotype before being re-infused into a properly
preconditioned patient with cancer.
Closing Remarks and Future Directions
Compared
to naturals DCs, aAPCs are proven to be a simpler and more cost-effective
method for expanding genetically engineered and antigen-specific T cells for
adoptive cellular therapy. aAPC platforms allow endless combinations of signal
1 and 2 for expanding the optimal T cell for specific malignancies. The
evolution of aAPC platforms bring clinicians one-step closer to harnessing the
power and ability of our own immune system to fight off even the most
detrimental diseases. Currently, researchers are discovering novel ways to
obtain robust T cell expansions of high quality and quantity by using various
inhibitory drugs and manipulations used in cell cultures. Preclinical studies
using the PI3Kd inhibitory drug, CAL-101, for
individuals with CLL, are being explored as a treatment modality [78], as well
as, supplementation for T cell cultures. Another alternative involves the use
of various costimulatory molecules on aAPCs. Researchers are exchanging CD28
for the costimulatory molecules ICOS or 41BB to explore potential T-cell
potency. Emerging studies are revealing the therapeutic effectiveness of Th17
cells in preclinical mouse models. A subset of CD4+ T cells once thought to be
a controversial lineage for cancer immunotherapies is now a potentially
advantageous subset for adoptive transfer due to their cytolytic capacity,
ability to have self-renewal properties, and ability to persist [79]. When
Th17, and even IL-17-producing CD8+ T cells (Tc17), are expanded with ICOS, their
antitumor efficacy increases compared to co-stimulation with CD28 [80,81].
Other emerging concepts in the world of aAPCs are the methods to enrich
autologous antigen-specific T-cells from cancer patients as a potential cell
transfer therapy. As described earlier, the nano-scale aAPC platform is a novel
approach to enrich antigen-specific T-cells with an HLA-A2+
antigen-presentation [73]. Further preclinical studies in our lab are
investigating the optimal signal 1, comparing dimers versus tetramers to enrich
antigen-specific T cells. Whether this approach can effectively expand
tumor-specific T cells with a less differentiated phenotype and maintain
functional capacity is yet to be known. The developments of aAPCs have been
improved significantly since the 1995 CD3/CD28 beads. Each aAPC provides
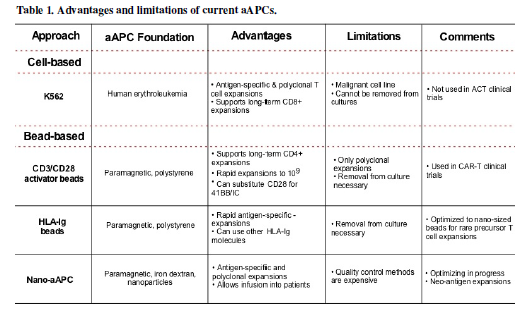
advantages over the other, as well as, limitations, as shown in Table 1. Further investigations are
underway to achieve optimal aAPC protocols to generate durable memory T cells
for broad use in the clinics.
ACKNOWLEDGEMENT AND FUNDING
This project is supported in part by funds from the Departments of Microbiology & Immunology, Hematology/Oncology, Surgery, and ACS Institutional Research Grant(JCV), K12(JCV), and NCI-1 RO1CA208514(CMP).
- Rosenberg SA, Dudley ME (2009) Adoptive
cell therapy for the treatment of patients with metastatic melanoma. Curr
Opin Immunol 21:233-240.
- Rosenberg SA, Dudley ME (2003)
Adoptive-cell-transfer therapy for the treatment of patients with cancer.
Nature reviews. Cancer 3: 666-675.
- Dudley ME, Wunderlich JR, Yang JC, Hwu
P, Schwartzentruber DJ, et al. (2002) A phase I study of nonmyeloablative
chemotherapy and adoptive transfer of autologous tumor antigen-specific T
lymphocytes in patients with metastatic melanoma. J Immunother 25:
243-251.
- Rosenberg SA, Packard BS, Aebersold PM,
Solomon D, Topalian SL, et al. (1988) Use of tumor-infiltrating
lymphocytes and interleukin-2 in the immunotherapy of patients with
metastatic melanoma. A preliminary report. N Engl J Med 319: 1676-1680.
- Maude SL, Teachey DT, Porter DL,
Grupp SA (2015) CD19-targeted
chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia.
Blood 125: 4017-4023.
- Kochenderfer JN, Wilson WH, Janik JE,
Dudley ME, Stetler-Stevenson M, et al. (2010) Eradication of B-lineage
cells and regression of lymphoma in a patient treated with autologous T
cells genetically engineered to recognize CD19. Blood 116:4099-4102.
- Amos SM, Pegram HJ, Westwood JA, John
LB, Devaud C, et al. (2011) Adoptive immunotherapy combined with
intratumoral TLR agonist delivery eradicates established melanoma in mice.
Cancer Immunol Immunother 60: 671-683.
- Kvistborg P, Shu CJ, Heemskerk B,
Fankhauser M, Thrue CA, et al. (2012) TIL therapy broadens the
tumor-reactive CD8(+) T cell compartment in melanoma patients. Oncoimmunol
1: 409-418.
- Phan GQ, Rosenberg SA (2013) Adoptive
cell transfer for patients with metastatic melanoma: the potential and
promise of cancer immunotherapy. Cancer Control 20: 289-297.
- Shannon
LM, Noelle F, Pamela AS, Richard A, David MB, et al. (2014)
Chimeric antigen receptor T cells for sustained remissions in leukemia. N
Engl J Med 371: 1507-1517.
- Lee DW, Kochenderfer JN,
Stetler-Stevenson M, Cui YK, Delbrook C, et al. (2015) T cells expressing
CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in
children and young adults: a phase 1 dose-escalation trial. Lancet 385:
517-528.
- Kalos M, Levine BL, Porter DL, Katz S,
Grupp SA, et al. (2011) T cells with chimeric antigen receptors have
potent antitumor effects and can establish memory in patients with
advanced leukemia. Sci Transl Medicine 3: 95ra73.
- Savoldo B, Ramos CA, Liu E, Mims MP,
Keating MJ, et al. (2011) CD28 costimulation improves expansion and
persistence of chimeric antigen receptor-modified T cells in lymphoma
patients. J Clin Invest 121: 1822-1826.
- Jensen MC, Popplewell L, Cooper LJ,
DiGiusto D, Kalos M, et al. (2010) Antitransgene rejection responses
contribute to attenuated persistence of adoptively transferred
CD20/CD19-specific chimeric antigen receptor redirected T cells in humans.
Biol Blood Marrow Transplant 16: 1245-1256.
- Rosenberg SA, Yang JC, White DE,
Steinberg SM (1998) Durability of complete responses in patients with
metastatic cancer treated with high-dose interleukin-2: identification of
the antigens mediating response. Ann Surg 228: 307-319.
- Dudley ME, Gross CA, Langhan MM, Garcia
MR, Sherry RM, et al. (2010) CD8+ enriched "young" tumor
infiltrating lymphocytes can mediate regression of metastatic melanoma.
Clin Cancer Res16: 6122-6131.
- Dudley ME, Wunderlich JR, Yang JC, Sherry
RM, Topalian SL, et al. (2005) Adoptive cell transfer therapy following
non-myeloablative but lymphodepleting chemotherapy for the treatment of
patients with refractory metastatic melanoma. J Clin Oncol 23: 2346-2357.
- Wrzesinski C, Paulos CM, Kaiser A,
Muranski P, Palmer DC, et al. (2010) Increased intensity lymphodepletion
enhances tumor treatment efficacy of adoptively transferred tumor-specific
T cells. J Immunother 33: 1-7.
- Yao X, Ahmadzadeh M, Lu YC, Liewehr DJ,
Dudley ME, et al. (2012) Levels of peripheral CD4(+)FoxP3(+) regulatory T
cells are negatively associated with clinical response to adoptive
immunotherapy of human cancer. Blood 119: 5688-5696.
- Gattinoni L, Finkelstein SE, Klebanoff
CA, Antony PA, Palmer DC, et al. (2005) Removal of homeostatic cytokine
sinks by lymphodepletion enhances the efficacy of adoptively transferred
tumor-specific CD8+ T cells. J Exp Med 202: 907-912.
- Nelson MH, Diven MA, Huff LW, Paulos CM
(2015) Harnessing the Microbiome to Enhance Cancer Immunotherapy. J
Immunol Res 368736.
- Busch DH, Frassle SP, Sommermeyer D,
Buchholz VR, Riddell SR (2016) Role of memory T cell subsets for adoptive
immunotherapy. Semin Immunol 28: 28-34.
- van der Windt GJ, Everts B, Chang CH,
Curtis JD, Freitas TC, et al. (2012) Mitochondrial respiratory capacity is
a critical regulator of CD8+ T cell memory development. Immunity 36:
68-78.
- D. O'Sullivan, E. L. Pearce, Targeting T
cell metabolism for therapy. Trends Immunol36, 71-80 (2015).
- Lanzavecchia A, Sallusto F (2002)
Progressive differentiation and selection of the fittest in the immune
response. Nature reviews. Immunology 2: 982-987.
- Gattinoni L, Klebanoff CA, Palmer DC,
Wrzesinski C, Kerstann K, et al. (2005) Acquisition of full effector
function in vitro paradoxically impairs the in vivo antitumor efficacy of
adoptively transferred CD8+ T cells. J Clin Invest 115: 1616-1626.
- Yang S, Gattinoni L, Liu F, Ji Y, Yu Z,
et al. (2011) In vitro generated anti-tumor T lymphocytes exhibit distinct
subsets mimicking in vivo antigen-experienced cells. Cancer Immunol
Immunother 60: 739-749.
- Wherry EJ, Teichgräber V, Becker TC,
Masopust D, Kaech SM, et al. (2003) Lineage relationship and protective
immunity of memory CD8 T cell subsets. Nature Immunol 4: 225-234.
- Turcotte S, Gros A, Hogan K, Tran E,
Hinrichs CS, et al. (2013) Phenotype and function of T cells infiltrating
visceral metastases from gastrointestinal cancers and melanoma:
implications for adoptive cell transfer therapy. J Immunol 191: 2217-2225.
- Sallusto F, Geginat J, Lanzavecchia A
(2004) Central memory and effector memory T cell subsets: function,
generation, and maintenance. Annu Rev Immunol 22: 745-763.
- Graef
P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, et al. (2014)
Serial transfer of single-cell-derived immunocompetence reveals stemness
of CD8(+) central memory T cells. Immunity 41: 116-126.
- Wang X, Popplewell LL, Wagner JR,
Naranjo A, Blanchard MS, et al. (2016) Phase 1 studies of central
memory-derived CD19 CAR T-cell therapy following autologous HSCT in
patients with B-cell NHL. Blood 127: 2980-2990.
- Purvis HA, Stoop JN, Mann J, Woods S,
Kozijn AE, et al. (2010) Low-strength T-cell activation promotes Th17
responses. Blood 116: 4829-4837.
- Alvarez-Fernandez C, Escriba-Garcia L, Vidal S, Sierra
J, Briones J (2016) A short CD3/CD28
costimulation combined with IL-21 enhance the generation of human memory
stem T cells for adoptive immunotherapy. J Transl Med 14: 214.
- Gett AV, Sallusto F, Lanzavecchia A,
Geginat J (2003) T cell fitness determined by signal strength. Nature
Immunol 4: 355-360.
- Colombetti S, Basso V, Mueller DL,
Mondino A (2006) Prolonged TCR/CD28 engagement drives IL-2-independent T
cell clonal expansion through signaling mediated by the mammalian target
of rapamycin. J Immunol 176: 2730-2738.
- Mackensen A, Herbst B, Chen JL, Köhler
G, Noppen C, et al. (2000) Phase I study in melanoma patients of a vaccine
with peptide-pulsed dendritic cells generated in vitro from CD34(+)
hematopoietic progenitor cells. Int J Cancer 86: 385-392.
- Poschke I, Lövgren T, Adamson L, Nyström
M, Andersson E, et al. (2014) A phase I clinical trial combining dendritic
cell vaccination with adoptive T cell transfer in patients with stage IV
melanoma. Cancer Immunol Immunother 63: 1061-1071.
- Almand B, Resser JR, Lindman B, Nadaf S,
Clark JI, et al. (2000) Clinical significance of defective dendritic cell
differentiation in cancer. Clin Cancer Res 6: 1755-1766.
- Ratta M, Fagnoni F, Curti A, Vescovini
R, Sansoni P, et al. (2002) Dendritic cells are functionally defective in
multiple myeloma: the role of interleukin-6. Blood 100: 230-237.
- Satthaporn S, Robins A, Vassanasiri W,
El-Sheemy M, Jibril JA, et al. (2004) Dendritic cells are dysfunctional in
patients with operable breast cancer. Cancer Immunol Immunother 53:
510-518.
- Steinman RM, Cohn ZA (1973)
Identification of a novel cell type in peripheral lymphoid organs of mice.
I. Morphology, quantitation, tissue distribution. J Exp Med 137:
1142-1162.
- Nieda M, Tomiyama M, Egawa K (2003) Ex
vivo enhancement of antigen-presenting function of dendritic cells and its
application for DC-based immunotherapy. Hum Cell 16: 199-204.
- Banchereau J, Steinman RM (1998)
Dendritic cells and the control of immunity. Nature 392: 245-252.
- Rossi M, Young JW (2005) Human dendritic
cells: potent antigen-presenting cells at the crossroads of innate and
adaptive immunity. J Immunol 175: 1373-1381.
- Inaba K, Metlay JP, Crowley MT, Steinman
RM (1990) Dendritic cells pulsed with protein antigens in vitro can prime
antigen-specific, MHC-restricted T cells in situ. J Exp Med 172: 631-640.
- Norbury CC, Hewlett LJ, Prescott AR,
Shastri N, Watts C (1995) Class I MHC presentation of exogenous soluble
antigen via macropinocytosis in bone marrow macrophages. Immunity 3:
783-791.
- Steinman RM (1991) The dendritic cell
system and its role in immunogenicity. Annu Rev Immunol 9: 271-296.
- Sallusto F, Lanzavecchia A (1994)
Efficient presentation of soluble antigen by cultured human dendritic
cells is maintained by granulocyte/macrophage colony-stimulating factor
plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp
Med 179: 1109-1118.
- Rissoan MC, Soumelis V, Kadowaki N,
Grouard G, Briere F, et al. (1999) Reciprocal control of T helper cell and
dendritic cell differentiation. Science 283: 1183-1186.
- Palucka K, Banchereau J (2012) Cancer
immunotherapy via dendritic cells. Nature reviews. Cancer 12: 265-277.
- Paczesny S, Banchereau J, Wittkowski KM,
Saracino G, Fay J, et al. (20004) Expansion of melanoma-specific cytolytic
CD8+ T cell precursors in patients with metastatic melanoma vaccinated
with CD34+ progenitor-derived dendritic cells. J Exp Med 199: 1503-1511.
- Dhodapkar MV, Steinman RM, Krasovsky J,
Munz C, Bhardwaj N (2001) Antigen-specific inhibition of effector T cell
function in humans after injection of immature dendritic cells. J Exp Med
193: 233-238.
- Chodon T, Comin-Anduix B, Chmielowski B,
Koya RC, Wu Z, et al. (2014) Adoptive transfer of MART-1 T-cell receptor
transgenic lymphocytes and dendritic cell vaccination in patients with
metastatic melanoma. Clin Cancer Res 20: 2457-2465.
- Butler MO, Hirano N (2014) Human
cell-based artificial antigen-presenting cells for cancer immunotherapy.
Immunol Rev 257: 191-209.
- Lozzio BB, Lozzio CB (1977) Properties
of the K562 cell line derived from a patient with chronic myeloid
leukemia. Int J Cancer 19: 136.
- Kim JV, Latouche JB, Riviere I, Sadelain
M (2004) The ABCs of artificial antigen presentation. Nat Biotechnol 22:
403-410.
- Butler MO, Lee JS, Ansén S, Neuberg D,
Hodi FS, et al. (2007) Long-lived antitumor CD8+ lymphocytes for adoptive
therapy generated using an artificial antigen-presenting cell. Clin Cancer
Res13: 1857-1867.
- Hirano N, Butler MO, Xia Z, Ansén S, von
Bergwelt-Baildon MS, et al. (2006) Engagement of CD83 ligand induces
prolonged expansion of CD8+ T cells and preferential enrichment for
antigen specificity. Blood 107: 1528-1536.
- Levine BL, Cotte J, Small CC, Carroll
RG, Riley JL, et al. (1998) Large-scale production of CD4+ T cells from
HIV-1-infected donors after CD3/CD28 costimulation. J Hematother 7: 437-448.
- Porter DL, Levine BL, Bunin N,
Stadtmauer EA, Luger SM, et al., (2006) A phase 1 trial of donor
lymphocyte infusions expanded and activated ex vivo via CD3/CD28
costimulation. Blood 107: 1325-1331.
- Levine BL, Bernstein WB, Connors M,
Craighead N, Lindsten T, et al. (1997)
Effects of CD28 costimulation on long-term proliferation of CD4+ T
cells in the absence of exogenous feeder cells. J Immunol 159: 5921-5930.
- Weng NP, Palmer LD, Levine BL, Lane HC,
June CH, et al. (1997) Tales of tails: regulation of telomere length and
telomerase activity during lymphocyte development, differentiation,
activation, and aging. Immunol Rev
160: 43-54.
- Hollyman D, Stefanski J, Przybylowski M,
Bartido S, Borquez-Ojeda O, et al. (2009) Manufacturing validation of biologically
functional T cells targeted to CD19 antigen for autologous adoptive cell
therapy. J Immunother 32: 169-180.
- Kochenderfer JN, Yu Z, Frasheri D,
Restifo NP, Rosenberg SA (2010) Adoptive transfer of syngeneic T cells
transduced with a chimeric antigen receptor that recognizes murine CD19
can eradicate lymphoma and normal B cells. Blood116, 3875-3886.
- Porter DL, Levine BL, Kalos M, Bagg A,
June CH (2011) Chimeric antigen receptor-modified T cells in chronic
lymphoid leukemia. N Engl J Med 365: 725-733.
- Zhang T, Cao L, Xie J, Shi N, Zhang Z,
et al. (2015) Efficiency of CD19 chimeric antigen receptor-modified T
cells for treatment of B cell malignancies in phase I clinical trials: a
meta-analysis. Oncotarget 6: 33961-33971.
- Thery C, Zitvogel L, Amigorena S (2002)
Exosomes: composition, biogenesis and function. Nat Rev Immunol 2:
569-579.
- Oelke M, Maus MV, Didiano D, June CH,
Mackensen A, et al. (2003) Ex vivo induction and expansion of
antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting
cells. Nat Med 9: 619-624.
- Oelke M, Schneck JP (2004) HLA-Ig-based
artificial antigen-presenting cells: setting the terms of engagement. Clin
Immunol 110: 243-251.
- Oelke M, Schneck JP (2010) Overview of a
HLA-Ig based "Lego-like system" for T cell monitoring,
modulation and expansion. Immunol Res 47: 248-256.
- Durai M, Krueger C, Ye Z, Cheng L,
Mackensen A, et al. (2009) In vivo
functional efficacy of tumor-specific T cells expanded using HLA-Ig based
artificial antigen presenting cells (aAPC). Cancer Immunol Immunother 58:
209-220.
- Perica K, Bieler JG, Schütz C, Varela
JC, Douglass J, et al. (2015) Enrichment and Expansion with Nanoscale
Artificial Antigen Presenting Cells for Adoptive Immunotherapy. ACS Nano
9: 6861-6871.
- Perica K, De León Medero A, Durai M,
Chiu YL, Bieler JG, et al. (2014) Nanoscale artificial antigen presenting
cells for T cell immunotherapy. Nanomedicine 10: 119-129.
- Fahmy TM, Bieler JG, Edidin M, Schneck
JP (2001) Increased TCR avidity after T cell activation: a mechanism for
sensing low-density antigen. Immunity 14: 135-143.
- Perica K, Tu A, Richter A, Bieler JG,
Edidin M, et al. (2014) Magnetic field-induced T cell receptor clustering
by nanoparticles enhances T cell activation and stimulates antitumor
activity. ACS Nano 8: 2252-2260.
- Jenkins MK, Moon JJ (2012) The role of
naive T cell precursor frequency and recruitment in dictating immune
response magnitude. J Immunol 188: 4135-4140.
- Hoellenriegel J, Meadows SA, Sivina M,
Wierda WG, Kantarjian H, et al. (2011) The phosphoinositide 3'-kinase
delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine
networks in chronic lymphocytic leukemia. Blood 118: 3603-3612.
- Muranski P, Borman ZA, Kerkar SP,
Klebanoff CA, Ji Y, et al. (2011) Th17 cells are long lived and retain a
stem cell-like molecular signature. Immunity 35: 972-985.
- Nelson MH, Kundimi S, Bowers JS, Rogers
CE, Huff LW, et al. (2015) The inducible costimulator augments Tc17 cell
responses to self and tumor tissue. J Immunol 194: 1737-1747.
- Paulos CM, Carpenito C, Plesa G, Suhoski
MM, Varela-Rohena A, et al. (2010) The inducible costimulator (ICOS) is
critical for the development of human T(H)17 cells. Sci Transl Medicine 2,
55ra78.
-
 Table 1
Table 1
QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- International Journal of AIDS (ISSN: 2644-3023)
- Oncology Clinics and Research (ISSN: 2643-055X)
- Dermatology Clinics and Research (ISSN:2380-5609)
- Journal of Renal Transplantation Science (ISSN:2640-0847)
- Journal of Cell Signaling & Damage-Associated Molecular Patterns
- Journal of Clinical Trials and Research (ISSN:2637-7373)
- International Journal of Surgery and Invasive Procedures (ISSN:2640-0820)



